
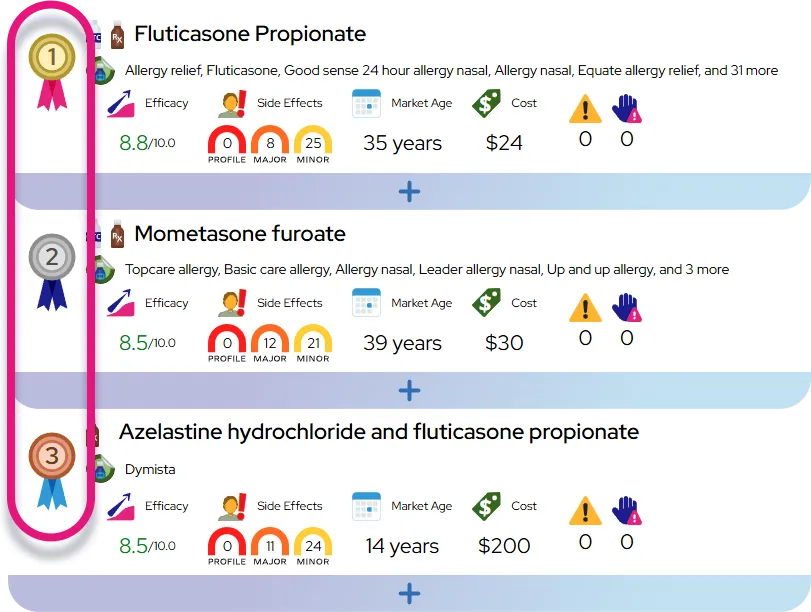
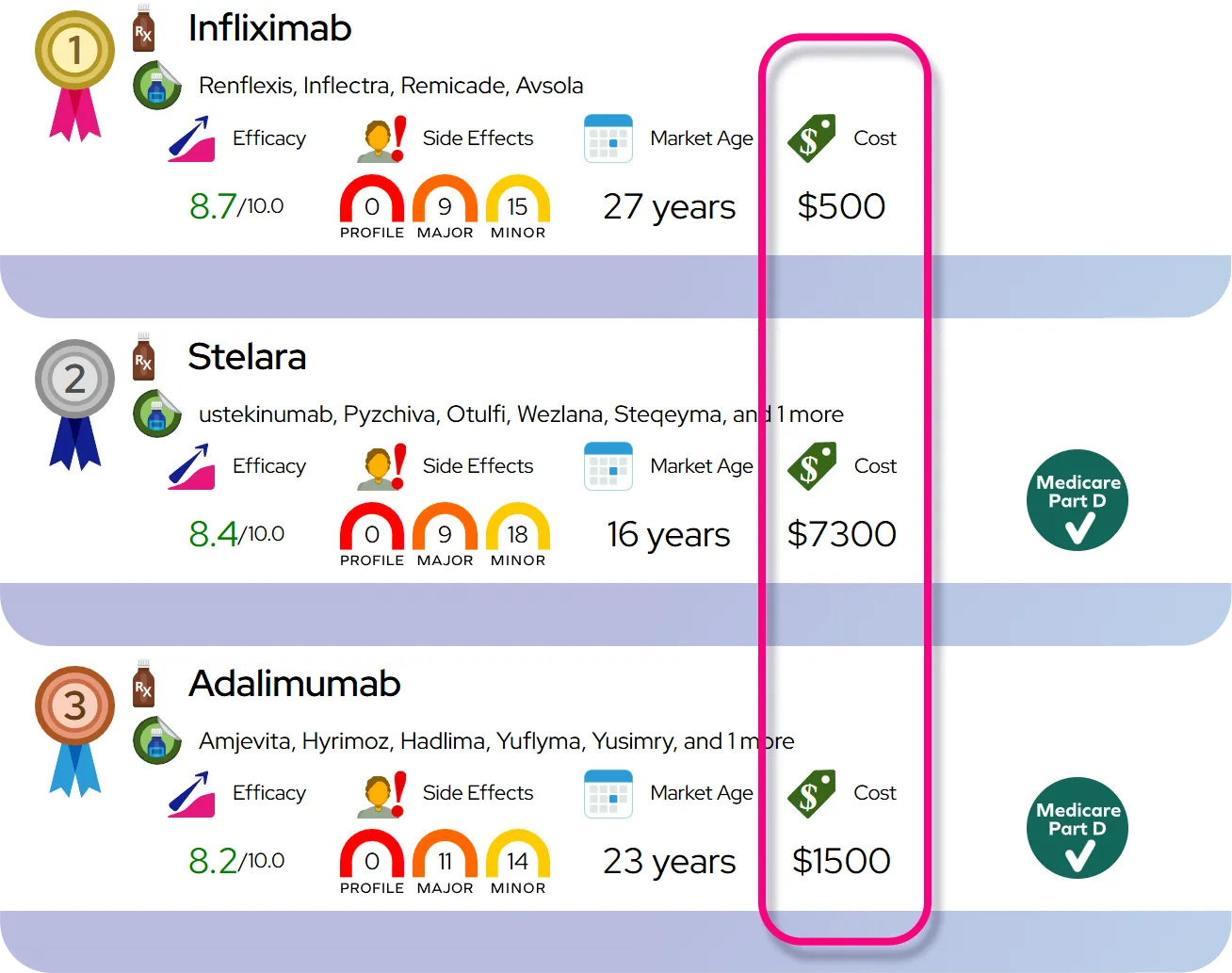
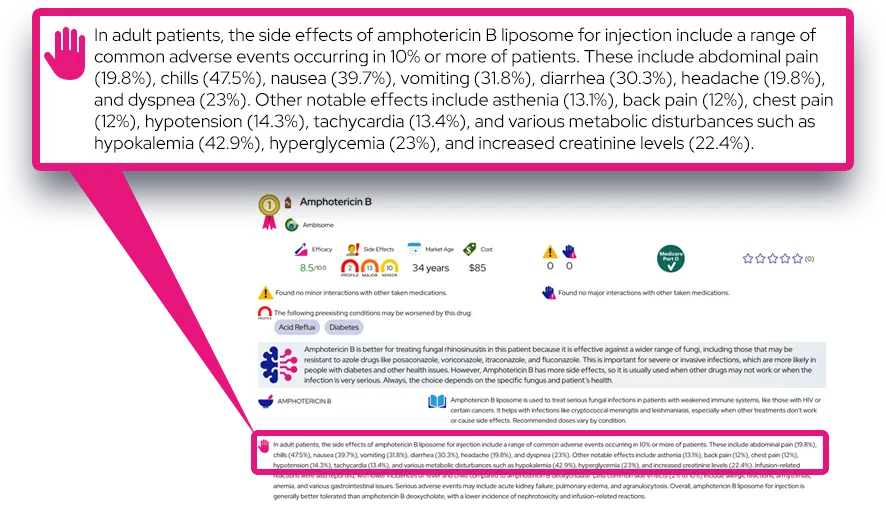
Alagille syndrome
At a Glance
 It is a chronic, lifelong condition that is generally manageable with medication and diet, although severe cases may require surgery or transplantation.
It is a chronic, lifelong condition that is generally manageable with medication and diet, although severe cases may require surgery or transplantation.











How It Affects You
Alagille syndrome is a multisystem genetic disorder that primarily affects the liver, heart, and other organs by disrupting normal development. The condition leads to a reduction in the number of bile ducts inside the liver, causing bile accumulation and potential scarring.
- Liver involvement results in jaundice, intense itching, and difficulty absorbing nutrients.
- Heart defects, such as pulmonary stenosis, are common and can impair blood flow.
- Other effects include distinctive facial features, kidney abnormalities, and skeletal changes like butterfly vertebrae.
Causes and Risk Factors
Causes
Alagille syndrome is caused by a genetic mutation, most commonly in the JAG1 gene and less frequently in the NOTCH2 gene. These genes regulate the Notch signaling pathway, which is essential for the proper formation of various body systems during fetal development. The errors in this pathway lead to the scarcity of bile ducts in the liver, as well as defects in the heart, spine, and other organs.
Risk Factors
The primary risk factor is family history, as the condition follows an autosomal dominant inheritance pattern. This means a child only needs to inherit one copy of the mutated gene from a parent to develop the syndrome. However, in more than half of all cases, the mutation occurs spontaneously (de novo) in the child, meaning parents without the condition can still have a child with Alagille syndrome.
Prevention
There is currently no known way to prevent Alagille syndrome since it is a genetic disorder determined at conception. Primary prevention strategies focus on genetic counseling for affected individuals or prospective parents with a known family history. This helps families understand the odds of passing the condition to future children, though it does not prevent the occurrence of spontaneous mutations.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
The symptoms of Alagille syndrome can range from very mild to severe, even among members of the same family. The most common sign is cholestasis, a reduction in bile flow, which causes jaundice (yellowing of the skin and eyes), pale stools, dark urine, and intense itching (pruritus). Children often experience failure to thrive and growth delays due to poor nutrient absorption. Distinctive facial features are also common, including a broad forehead, deep-set eyes, and a pointed chin. Other signs include heart murmurs caused by narrowed pulmonary arteries, skeletal abnormalities like "butterfly vertebrae" seen on X-rays, and fatty skin deposits called xanthomas.
Diagnosis
Doctors diagnose the condition based on a combination of clinical symptoms and test results. A liver biopsy is frequently performed to confirm a paucity (shortage) of bile ducts. Genetic testing can identify mutations in the JAG1 or NOTCH2 genes. Additional diagnostic tools include an echocardiogram to detect heart defects, an abdominal ultrasound to examine the liver and kidneys, and a specialized eye exam to look for posterior embryotoxon, a specific abnormality on the surface of the eye.
Treatment and Management
Medications and Nutrition
Treatment mainly focuses on increasing bile flow and managing symptoms. Doctors often prescribe ursodiol to help move bile through the liver. Because the condition prevents the absorption of dietary fats, patients require high-calorie diets and supplements of fat-soluble vitamins (A, D, E, and K). Medications such as antihistamines or rifampin may be used to control severe itching, which can significantly impact quality of life.
Procedures and Surgery
If medications are ineffective, surgical procedures may be considered. Biliary diversion surgery can help lower the amount of bile acids in the blood, thereby reducing itching and xanthomas. In cases of severe liver failure or cirrhosis, a liver transplant may be the best option. Heart defects associated with the syndrome may also require surgical repair or catheter-based interventions.
When to Seek Medical Care
Regular monitoring by a specialist is crucial. You should see a doctor if you notice signs of worsening liver function, such as increased jaundice, unexplained bleeding or bruising, or sudden abdominal swelling. Emergency care is necessary if the patient experiences vomiting of blood, bloody stools, or severe lethargy.
Severity and Prognosis
Severity and Disease Course
Alagille syndrome is characterized by variable expressivity, meaning the severity differs greatly from person to person. Some individuals have only minor symptoms that do not require treatment, while others face life-threatening organ failure in childhood. The liver disease typically presents early but often stabilizes or even improves as the child grows older, though the risk of complications remains.
Complications
Major complications include chronic malnutrition and growth failure due to the inability to absorb fat. Long-term risks involve progressive liver scarring (cirrhosis) and portal hypertension. Cardiovascular complications, such as narrowing of the pulmonary arteries or Tetralogy of Fallot, can also be severe. Less common but serious risks include intracranial bleeding due to blood vessel abnormalities and kidney dysfunction.
Prognosis
With early diagnosis and proper management, the prognosis for most people with Alagille syndrome is favorable. Life expectancy is generally normal for those who do not suffer from severe heart defects or liver failure. However, mortality rates are higher in patients with complex heart disease or intracranial bleeding. Advances in liver transplantation have significantly improved survival rates for those with severe liver involvement.
Impact on Daily Life
Daily Living and Coping
Living with Alagille syndrome requires managing a strict regimen of medications and dietary supplements. Children may need support at school to manage fatigue or medical appointments. The visible signs of the disease, such as jaundice, xanthomas, or smaller stature, can affect self-esteem and social interactions. Coping strategies include connecting with support groups and ensuring open communication with teachers and peers about the condition.
Questions to Ask Your Healthcare Provider
Being prepared for appointments can help manage the condition effectively. Consider asking the following questions:
- What are the specific signs that my child's liver disease might be progressing?
- Are there any specific sports or physical activities that should be avoided?
- How can we best manage the intense itching if current medications aren't working?
- What is the long-term plan for monitoring heart and kidney function?
- Should other family members undergo genetic testing?
Common Questions and Answers
Q: Is Alagille syndrome contagious?
A: No, it is a genetic disorder and cannot be spread from person to person.
Q: Will my child grow out of Alagille syndrome?
A: The condition is lifelong and genetic, so a child will not grow out of it. However, liver symptoms often stabilize or improve as the child gets older.
Q: Can people with Alagille syndrome have children?
A: Yes, many adults with the condition have children. Because it is a dominant genetic trait, there is a 50% chance of passing the condition to each child, so genetic counseling is often recommended.
Q: Do all patients need a liver transplant?
A: No. Only about 15% to 20% of children with Alagille syndrome develop severe liver failure or intractable symptoms that require a transplant.