
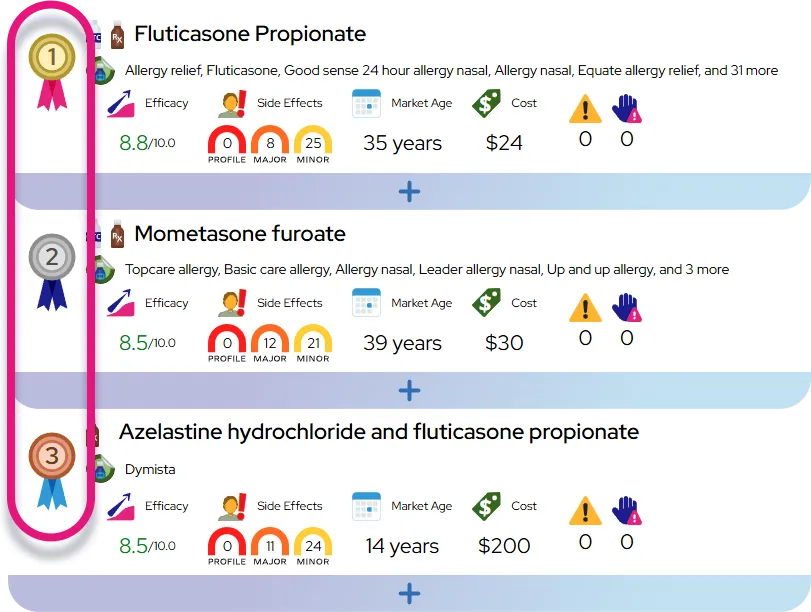
Atypical hemolytic uremic syndrome
At a Glance
 It is a chronic, lifelong condition driven by a genetic predisposition, characterized by the risk of sudden, acute episodes of disease activity that require immediate medical intervention.
It is a chronic, lifelong condition driven by a genetic predisposition, characterized by the risk of sudden, acute episodes of disease activity that require immediate medical intervention.










How It Affects You
Atypical hemolytic uremic syndrome (aHUS) is a systemic disease where the immune system attacks the lining of blood vessels, causing inflammation and the formation of small blood clots throughout the body. While this process most severely impacts the kidneys, the widespread nature of the blood vessel damage means it can affect multiple organ systems simultaneously.
- The condition leads to thrombotic microangiopathy, resulting in low platelet counts and the mechanical destruction of red blood cells.
- Blood clots restrict flow to vital organs, frequently causing acute kidney failure and high blood pressure.
- Extra-renal complications can occur, affecting the heart, brain, lungs, and gastrointestinal tract.
Causes and Risk Factors
Underlying Causes
Atypical hemolytic uremic syndrome is primarily caused by a dysregulation of the complement system, a part of the body's immune defense that normally destroys infection. In patients with this condition, the complement system becomes overactive and attacks the body's own healthy cells, specifically the endothelial cells lining the blood vessels. This overactivation is often linked to inherited genetic mutations in specific genes that regulate the complement system, such as CFH, CFI, or MCP, or the development of autoantibodies that block these regulatory proteins.
Triggers and Risk Factors
While genetic predisposition is the primary driver, a triggering event is often required to activate the disease. Common triggers that stress the immune system include viral or bacterial infections (such as the flu or upper respiratory infections), pregnancy or childbirth, certain medications (including some chemotherapy or immunosuppressive drugs), organ transplantation, and malignancies. Not everyone with a genetic mutation will develop the disease, suggesting that the combination of genetics and an environmental trigger is necessary for the condition to manifest.
Prevention
There is currently no way to prevent the underlying genetic susceptibility to this condition. Primary prevention focuses on avoiding known triggers where possible, though many triggers like common infections are difficult to avoid completely. For individuals with a known diagnosis, adherence to prescribed treatments is critical to prevent disease recurrence and minimize organ damage. Family members of diagnosed patients may undergo genetic counseling and screening to understand their own risk profile.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
The symptoms of atypical hemolytic uremic syndrome often appear suddenly and can be severe. The classic triad of clinical signs includes hemolytic anemia (destruction of red blood cells), thrombocytopenia (low platelet count), and acute kidney injury. Patients may experience extreme fatigue, pale skin (pallor), and shortness of breath due to anemia. Low platelets can cause easy bruising, small red spots on the skin (petechiae), or prolonged bleeding. Kidney involvement typically presents as decreased urine output, swelling in the legs or feet (edema), and high blood pressure. Because blood clots can form anywhere, patients may also experience confusion, seizures, chest pain, or digestive issues if the brain, heart, or gut are affected.
Diagnostic Tests
Diagnosis requires a combination of blood tests and the exclusion of other similar diseases. Physicians look for evidence of thrombotic microangiopathy, which includes a complete blood count showing low hemoglobin and platelets, and a blood smear revealing schistocytes (fragmented red blood cells). Kidney function tests will typically show elevated creatinine and urea levels. A critical part of diagnosis is ruling out other causes of microangiopathy; this involves testing for Shiga toxin to rule out infection-associated HUS (STEC-HUS) and checking ADAMTS13 enzyme activity levels to rule out thrombotic thrombocytopenic purpura (TTP).
Genetic Evaluation
Once a clinical diagnosis is suspected or confirmed, genetic testing is often performed to identify specific mutations in the complement system. While this helps confirm the diagnosis and predict the risk of recurrence or transplant outcomes, treatment is usually started immediately based on clinical presentation rather than waiting for genetic results, as these can take weeks to return.
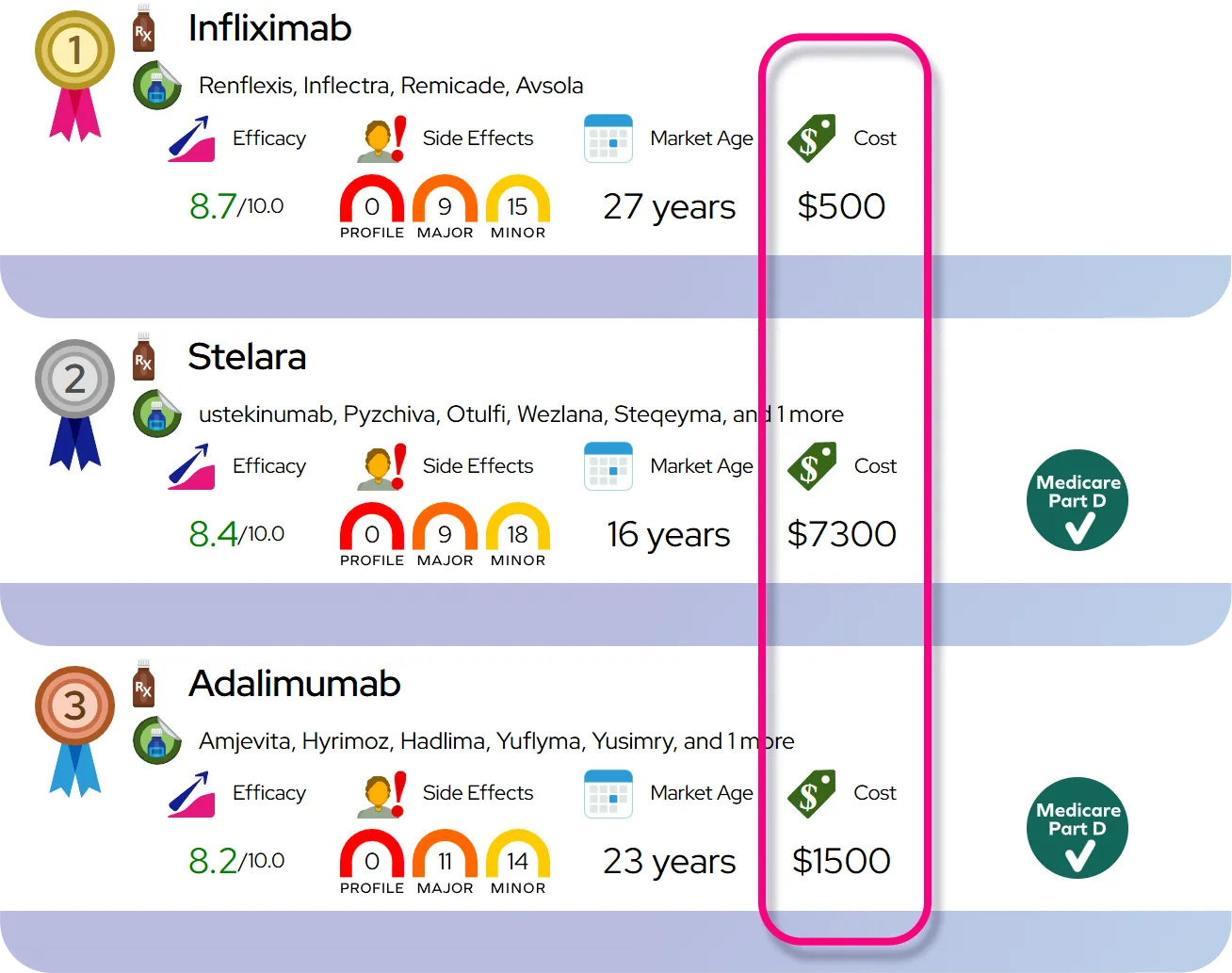
Treatment and Management
Medications
The standard of care for atypical hemolytic uremic syndrome involves the use of complement inhibitors, such as eculizumab or ravulizumab. These monoclonal antibodies work by blocking the specific part of the immune system (terminal complement pathway) that causes the uncontrolled attack on blood vessels. These medications are delivered via intravenous infusion and have revolutionized the management of the disease, often halting the destruction of blood cells and recovering kidney function.
Procedures and Supportive Care
Before the availability of complement inhibitors, plasma exchange or plasma infusion was the primary treatment. While plasma therapy may still be used in emergencies or if complement inhibitors are unavailable, it is generally less effective for treating the underlying complement dysregulation in many patients. Supportive care is also essential and may include red blood cell transfusions for severe anemia, platelet transfusions (only in life-threatening bleeding), and antihypertensive medications to manage high blood pressure.
Dialysis and Transplantation
Patients who experience severe kidney failure may require dialysis to filter waste from their blood. For those who progress to end-stage renal disease, a kidney transplant may be an option. However, kidney transplantation carries a high risk of the disease recurring in the new kidney unless the patient is treated with prophylactic complement inhibitors.
When to See a Doctor
Immediate medical attention is required if symptoms suggestive of a flare-up occur. Seek emergency care for signs such as sudden decrease in urine output, dark or tea-colored urine, unexplained bruising or bleeding, confusion, seizures, or extreme fatigue. Patients diagnosed with this condition require routine follow-up with a nephrologist or hematologist to monitor blood counts and kidney function, usually every few weeks or months depending on their treatment stability.
Severity and Prognosis
Severity and Disease Course
Atypical hemolytic uremic syndrome is a severe, high-stakes medical condition. Without timely treatment, the active phase of the disease can cause rapid and irreversible damage to the kidneys and other organs. The disease course is typically chronic, meaning patients remain at risk for relapses throughout their lives, especially when their immune system is stressed by infections or other triggers.
Prognosis and Life Expectancy
The prognosis has improved dramatically with the introduction of complement inhibitor therapy. Prior to these treatments, a significant percentage of patients progressed to end-stage kidney failure or died during the first acute episode. Today, with early diagnosis and access to appropriate medication, most patients can recover kidney function and live a normal lifespan. The risk of mortality is significantly reduced when the condition is managed effectively.
Complications
Long-term complications are primarily related to the extent of organ damage sustained before treatment stabilizes the condition. Chronic kidney disease (CKD) is a common long-term effect, potentially requiring lifelong dietary management or dialysis. High blood pressure is also a frequent complication that contributes to cardiovascular risk. In rare cases, if blood clots affected the brain or heart during an acute episode, patients may experience lasting neurological or cardiovascular issues.
Impact on Daily Life
Impact on Daily Activities
Living with this condition involves adapting to a routine of regular medical appointments and treatments. Patients on complement inhibitor therapy must receive intravenous infusions at regular intervals (e.g., every two to eight weeks), which can disrupt work or school schedules. Fatigue is a common complaint, even when the disease is well-managed, requiring patients to prioritize rest and energy conservation. Strict adherence to infection prevention protocols is also necessary, as the treatment suppresses part of the immune system, increasing susceptibility to certain infections like meningococcal disease.
Emotional and Mental Health
The unpredictability of flares and the chronic nature of the illness can lead to anxiety and stress. Patients may worry about the potential for kidney failure or the genetic implications for their children. Support groups and counseling can be valuable resources for navigating the emotional burden of a rare chronic disease.
Questions to Ask Your Healthcare Provider
Patients are encouraged to ask specific questions to better understand their management plan:
- What is my specific genetic mutation, and how does it affect my risk of relapse?
- How long will I need to remain on complement inhibitor therapy?
- What specific signs should prompt me to seek immediate emergency care?
- Are there any vaccinations I need to take or avoid while on treatment?
- What are the risks and benefits of kidney transplantation if my kidneys fail?
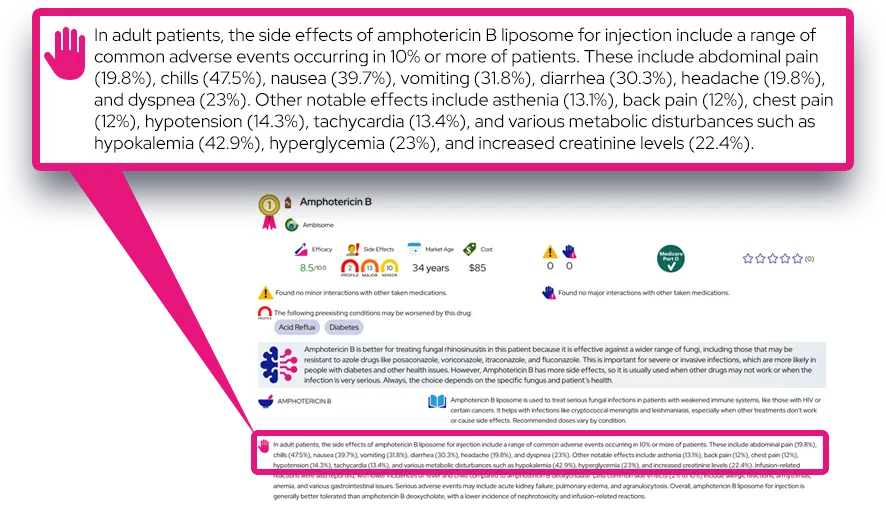
Common Questions and Answers
Q: Is atypical hemolytic uremic syndrome the same as the HUS caused by E. coli?
A: No, they are different conditions. The more common form, STEC-HUS, is caused by a bacterial infection from food poisoning, while atypical HUS is primarily caused by a genetic dysregulation of the immune system and is not caused by a digestive infection.
Q: Can this condition be cured permanently?
A: There is currently no permanent cure for the genetic predisposition underlying the disease. However, continuous treatment can effectively manage the condition, prevent symptoms, and protect organs from damage.
Q: Is it safe to get pregnant if I have this condition?
A: Pregnancy is a known trigger for disease flare-ups. Women with this condition should consult their specialist for preconception counseling to manage risks, as close monitoring and treatment adjustment are essential for a safe pregnancy.
Q: Do all family members with the gene get the disease?
A: No, having a genetic mutation does not guarantee you will develop the disease. This is known as incomplete penetrance, meaning other factors or triggers are usually needed to activate the condition.
Q: Why do I need the meningococcal vaccine?
A: The medications used to treat this condition block a specific part of the immune system that fights meningitis-causing bacteria. Vaccination provides critical protection against this serious infection while you are on treatment.