
Beta thalassemia
At a Glance
 This is a chronic, lifelong condition that requires ongoing management but is treatable and, in some cases, curable with advanced medical procedures.
This is a chronic, lifelong condition that requires ongoing management but is treatable and, in some cases, curable with advanced medical procedures.










How It Affects You
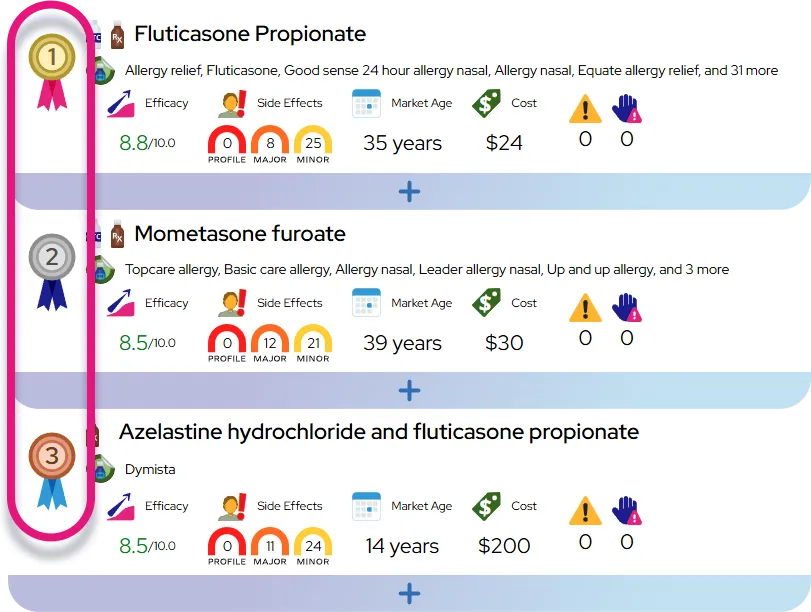
Beta thalassemia significantly impacts the body by impairing the production of hemoglobin, the protein in red blood cells responsible for carrying oxygen to tissues. This lack of oxygen causes the heart to work harder, often leading to exhaustion and weakness, while the bone marrow expands in an effort to produce more blood cells, potentially weakening bone structure. The condition also places strain on internal organs. Key effects on the body include:
- Systemic oxygen deprivation resulting in chronic fatigue and delayed growth in children.
- Changes in bone density and shape, particularly in the face and skull, due to bone marrow expansion.
- Risk of organ damage, specifically to the liver, heart, and spleen, often caused by iron overload from necessary blood transfusions.
Causes and Risk Factors
Biological Causes
Beta thalassemia is caused by mutations in the HBB gene, which provides instructions for making beta-globin, a component of hemoglobin. Hemoglobin is the red protein responsible for transporting oxygen in the blood. When these genes are altered, the body produces reduced amounts of beta-globin or none at all. This leads to a shortage of healthy red blood cells and a lack of oxygen reaching the body's tissues.
Genetic Inheritance
The condition is inherited in an autosomal recessive pattern, meaning a child must inherit two altered genes, one from each parent, to develop the moderate or severe forms of the disease. Individuals who inherit only one altered gene are carriers, often called beta thalassemia trait or minor, and usually do not have severe symptoms but can pass the gene to their children.
Risk Factors
The primary risk factor is a family history of the condition. Ancestry plays a significant role, as the genetic mutations are more common in people of Mediterranean, Middle Eastern, African, and Southeast Asian descent. There are no environmental or lifestyle risk factors that cause the condition, as it is strictly genetic.
Prevention and Screening
Because beta thalassemia is genetic, it cannot be prevented in the traditional sense of vaccines or lifestyle changes. However, genetic counseling and carrier screening are available for individuals with a family history or from high-risk ethnic groups who are planning to have children. These screenings can identify carriers and help prospective parents understand the risks of passing the condition to their offspring.

Diagnosis, Signs, and Symptoms
Common Signs and Symptoms
Symptoms vary significantly depending on the severity of the condition. Infants with the severe form often show signs within the first two years of life. Common symptoms include pale or yellowish skin (jaundice), poor appetite, dark urine, and fussiness. As the child grows, parents may notice delayed growth and development. In untreated or poorly managed cases, the physical stress of trying to produce red blood cells can cause the bones of the face to change shape and the abdomen to swell due to an enlarged spleen.
Diagnosing the Condition
Clinicians typically diagnose beta thalassemia through blood tests. A Complete Blood Count (CBC) usually reveals anemia and red blood cells that are smaller than normal. Hemoglobin electrophoresis is a specific test used to measure the types of hemoglobin in the blood and identify abnormal patterns. DNA testing may also be performed to confirm the diagnosis and identify the specific genetic mutations involved.
Differential Diagnosis
Doctors must distinguish beta thalassemia from other causes of anemia, most notably iron deficiency anemia. Because both conditions cause small, pale red blood cells, they can look similar on initial tests. However, treating beta thalassemia with iron supplements can be dangerous, so accurate diagnosis via hemoglobin studies and iron level testing is essential to rule out simple iron deficiency.
Treatment and Management
Medical Treatments
The main treatment for moderate to severe beta thalassemia is regular blood transfusions, which provide the body with the healthy red blood cells it cannot make on its own. While transfusions are lifesaving, they lead to a buildup of iron in the body. To manage this, patients undergo chelation therapy, which uses medications to remove excess iron and prevent organ damage. Folic acid supplements are often prescribed to help the body build new red blood cells.
Curative Options
A stem cell or bone marrow transplant is currently the only established cure for beta thalassemia. This procedure involves replacing the patient's defective blood-forming cells with healthy ones from a compatible donor. More recently, gene therapy has emerged as a potential treatment, where the patient's own stem cells are modified to produce normal hemoglobin.
Surgical Interventions
In some cases, the spleen may become enlarged and remove too many blood cells, increasing the need for transfusions. A surgery to remove the spleen (splenectomy) may be recommended, although this is done less frequently now due to the risk of infection.
Lifestyle and Monitoring
Patients generally need to avoid iron supplements unless a doctor confirms a deficiency. Eating a healthy diet and staying up to date with vaccines is important, especially if the spleen has been removed. Regular monitoring of iron levels, heart function, and liver health is crucial to manage the side effects of chronic transfusions.
When to Seek Medical Care
Patients should see a doctor immediately if they experience signs of infection, such as a high fever, especially if they have had their spleen removed. Worsening fatigue, shortness of breath, or chest pain also warrant prompt medical attention. Routine follow-up appointments are essential for monitoring transfusion schedules and iron levels.
Severity and Prognosis
Levels of Severity
Beta thalassemia is classified into three main types based on severity. Beta thalassemia minor (trait) is mild and often causes no symptoms. Beta thalassemia intermedia causes moderate anemia that may require occasional transfusions. Beta thalassemia major (Cooley's anemia) is the most severe form, requiring lifelong regular transfusions to survive.
Complications and Long-Term Effects
The primary complications stem from the disease itself and the iron overload caused by frequent blood transfusions. Excess iron can deposit in the heart, liver, and endocrine glands, leading to heart failure, liver disease, diabetes, or hormonal imbalances. The expansion of bone marrow can lead to thin, brittle bones (osteoporosis) and skeletal deformities.
Prognosis and Life Expectancy
Historically, severe beta thalassemia led to a shortened life expectancy. However, with consistent blood transfusions and effective iron chelation therapy, the outlook has improved dramatically. Many individuals with the severe form now live into their 40s, 50s, and beyond, leading productive lives. Early diagnosis and strict adherence to iron management plans are the most significant factors influencing a positive long-term outcome.
Impact on Daily Life
Impact on Activities and Emotional Health
Living with beta thalassemia requires adjusting to a schedule of regular medical appointments and treatments. Fatigue can sometimes interfere with school, work, or sports, requiring rest breaks or modified schedules. The chronic nature of the condition can also impact emotional well-being, making support from family, counselors, or patient advocacy groups valuable for coping with the stress of a lifelong illness.
Questions to Ask Your Healthcare Provider
Preparing questions for doctor visits can help patients manage their care effectively. Consider asking:
- What specific type of beta thalassemia do I (or my child) have?
- How often will blood transfusions be needed, and what is the schedule?
- What are the best options for managing iron overload?
- Are there any specific dietary restrictions I should follow?
- Am I a candidate for stem cell transplant or gene therapy?
- What signs of complications should I watch for at home?
Common Questions and Answers
Q: Is beta thalassemia contagious?
A: No, beta thalassemia is a genetic disorder. You cannot catch it from another person like a cold or flu; it is inherited from your parents.
Q: Can I take iron supplements to help with the anemia?
A: Generally, no. Unlike iron deficiency anemia, beta thalassemia often involves too much iron in the body, especially with transfusions. Taking extra iron can be dangerous and damage organs.
Q: Is there a cure for beta thalassemia?
A: Yes, a stem cell or bone marrow transplant can cure the condition, but it carries risks and requires a matching donor. Gene therapy is also becoming an option for some patients.
Q: Can women with beta thalassemia have children?
A: Yes, many women with beta thalassemia have healthy pregnancies. However, pregnancy requires close monitoring by specialists to manage anemia and cardiac health.
Q: Does beta thalassemia affect diet?
A: People with this condition are often advised to limit iron-rich foods, such as red meat. Drinking tea with meals may help reduce iron absorption, but specific dietary plans should be discussed with a doctor.