
Hereditary orotic aciduria
At a Glance
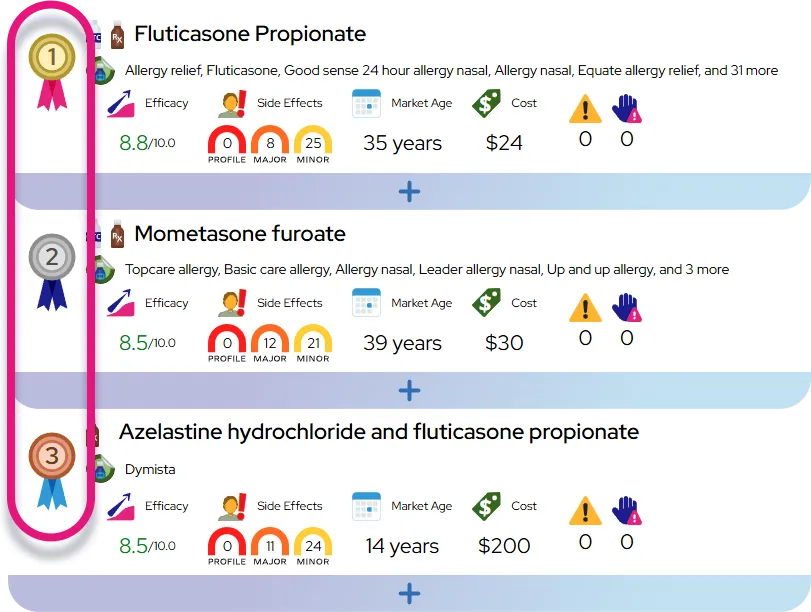
 It is a chronic, lifelong condition that is highly treatable and manageable with daily oral medication.
It is a chronic, lifelong condition that is highly treatable and manageable with daily oral medication.










How It Affects You
Hereditary orotic aciduria is a rare genetic disorder that affects the body's ability to produce uridine, a molecule essential for creating DNA and RNA. This metabolic defect has systemic effects, primarily targeting rapidly dividing cells in the bone marrow and impacting overall growth. Key effects on the body include:
- Severe anemia resulting in pale skin, fatigue, and weakness
- Formation of crystals in the urinary tract due to excess orotic acid excretion
- Delayed physical growth and slowed mental development in infants
Causes and Risk Factors
Underlying Biological Causes
Hereditary orotic aciduria is caused by mutations in the UMPS gene. This gene provides instructions for making an enzyme called uridine monophosphate synthase. This enzyme is crucial for the synthesis of pyrimidines, which are building blocks of DNA and RNA. When this enzyme is defective or missing, the body cannot produce uridine, leading to a shortage of nucleotides necessary for cell division and growth. Simultaneously, a substance called orotic acid builds up in the body because it cannot be converted into the next molecule in the metabolic pathway.
Genetic Risk Factors
The primary risk factor for this condition is family history, specifically having parents who are carriers of the gene mutation. The disorder follows an autosomal recessive inheritance pattern. This means a child must inherit two non-working copies of the gene, one from each parent, to develop the condition. Parents who carry only one copy of the mutated gene typically do not show symptoms but have a 25 percent chance of passing the condition to their children with each pregnancy.
Prevention Strategies
There is no known way to prevent the genetic mutation that causes hereditary orotic aciduria. For families with a known history of the disorder, genetic counseling is recommended to understand the risks of passing the condition to future children. Prenatal testing can determine if a fetus has inherited the condition, allowing for early preparation and management strategies immediately after birth.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms typically appear between one and three months of age. The most prominent sign is megaloblastic anemia, a blood disorder characterized by large, immature red blood cells that cannot function properly. This leads to pale skin, extreme tiredness, and lethargy. Infants often fail to gain weight or grow at the expected rate, a condition known as failure to thrive. Another specific symptom is the presence of orotic acid crystals in the urine, which may appear as cloudy urine or leave orange, sand-like sediment in diapers. Without treatment, children may experience significant physical and intellectual developmental delays.
Diagnostic Tests
Healthcare providers usually suspect this condition when a child has severe anemia that does not improve with standard vitamin B12 or folate supplements. Diagnosis is confirmed through specific laboratory tests. A urine analysis will reveal abnormally high levels of orotic acid. Blood tests are used to examine the size and count of blood cells. Genetic testing is the definitive method for diagnosis, identifying the specific mutations in the UMPS gene.
Differential Diagnosis
Hereditary orotic aciduria is often initially confused with other causes of megaloblastic anemia, such as vitamin B12 deficiency or folate deficiency. It may also be mistaken for other metabolic disorders like ornithine transcarbamylase deficiency, which also causes high levels of orotic acid but is accompanied by high ammonia levels in the blood, a symptom not found in hereditary orotic aciduria.
Treatment and Management
Medications and Replacement Therapy
The cornerstone of treatment is uridine replacement therapy. Patients are prescribed oral supplements of uridine (often in the form of uridine triacetate) to bypass the metabolic block. By providing the body with the uridine it cannot make on its own, the body can resume normal DNA and RNA synthesis. This medication addresses the root cause of the symptoms rather than just managing them. Treatment is typically lifelong and requires strict adherence to the dosing schedule.
Monitoring and Follow-up
Regular medical appointments are essential to monitor the effectiveness of the treatment. Doctors will track the child's growth, development, and blood counts to ensure the anemia has resolved. They will also monitor orotic acid levels in the urine. As a child grows, medication dosages often need adjustment based on weight.
When to Seek Medical Care
Parents should contact their healthcare provider if they notice any return of symptoms, such as:
- Renewed signs of fatigue or paleness
- Cloudy or orange sediment in the urine
- Difficulty urinating or signs of urinary tract pain
- Unexplained vomiting or lethargy
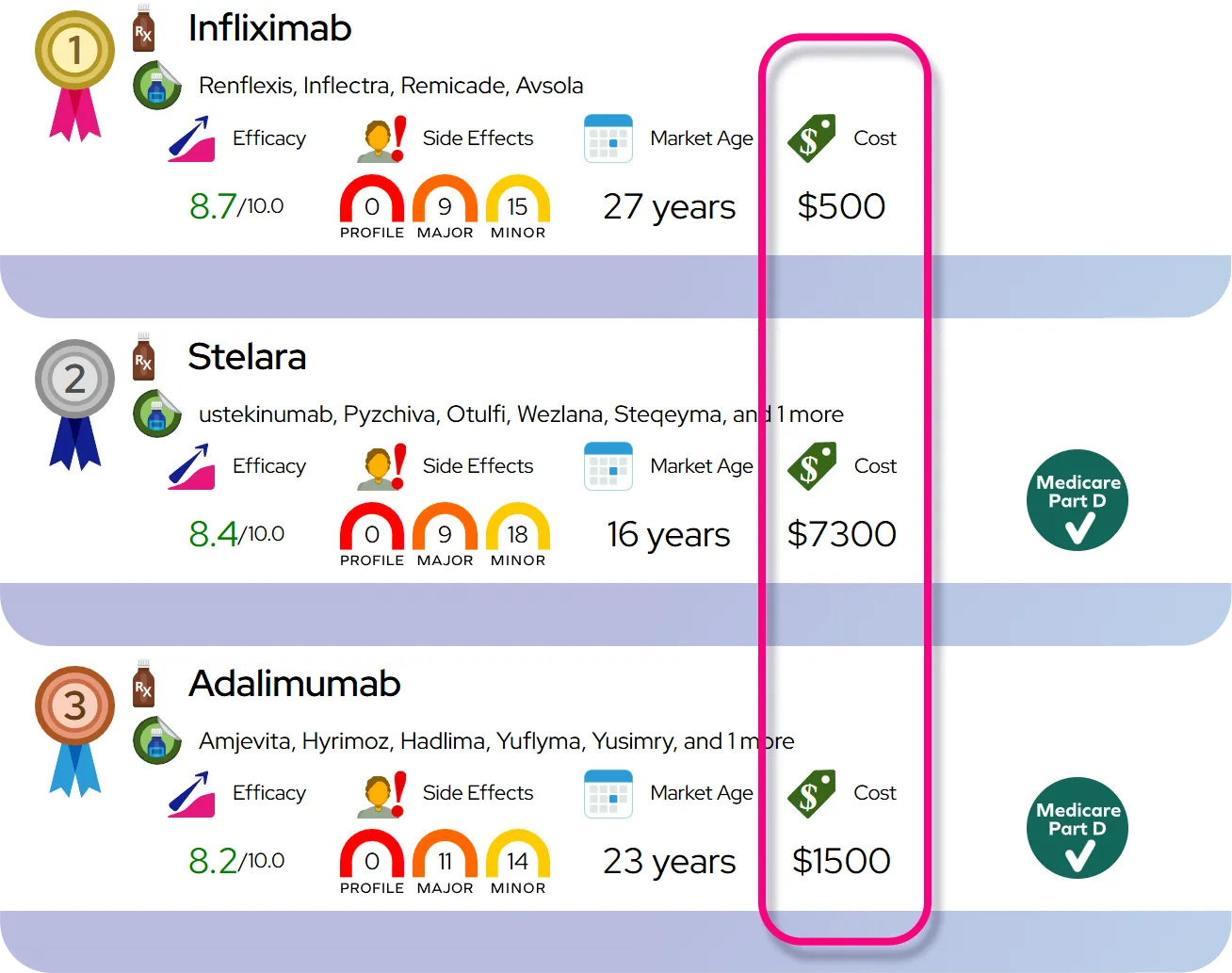
Severity and Prognosis
Severity and Disease Course
Untreated hereditary orotic aciduria is a severe and potentially fatal condition due to profound anemia and immune deficiency. However, with the advent of uridine replacement therapy, the severity is drastically reduced. The condition transforms from a life-threatening illness into a manageable chronic disorder. Symptoms like anemia usually resolve within a few weeks of starting treatment, and crystal excretion diminishes shortly thereafter.
Prognosis and Life Expectancy
The prognosis for treated individuals is excellent. When treatment is started early, children generally catch up on growth and developmental milestones, reaching normal height and weight. Life expectancy is considered normal for patients who remain on their medication. Long-term complications are rare in well-managed cases, although compliance with daily medication is critical to prevent relapse.
Impact on Daily Life
Daily Activities and Coping
Children and adults with treated hereditary orotic aciduria can participate fully in school, work, and recreational activities. There are typically no physical limitations once the anemia resolves. The primary impact on daily life is the routine of taking medication several times a day. Families may find it helpful to use alarms or pill organizers to ensure doses are not missed. Support groups for rare metabolic disorders can provide emotional support and practical advice for navigating the healthcare system.
Questions to Ask Your Healthcare Provider
- What is the correct dosage of uridine for my child's current weight?
- How frequently do we need to schedule follow-up blood and urine tests?
- Are there any side effects of the medication we should watch for?
- Does this condition require any specific dietary changes or restrictions?
- What information should I provide to school nurses or teachers about this condition?
Common Questions and Answers
Q: Is hereditary orotic aciduria contagious?
A: No, it is a genetic condition inherited from parents and cannot be spread from person to person.
Q: Can a child outgrow this condition?
A: No, the genetic defect is permanent, so the body will always need uridine supplements to function correctly.
Q: Does the treatment cure the developmental delays?
A: If treatment is started early, most developmental delays are reversible, and the child can catch up to their peers.
Q: Why didn't vitamins fix the anemia?
A: While the anemia looks like a vitamin deficiency, it is actually caused by the body's inability to make DNA building blocks, which is why only uridine replacement works.