
Homozygous familial hypercholesterolemia
At a Glance
 It is a severe, chronic condition that requires lifelong, intensive medical treatment to manage cholesterol levels and prevent fatal complications.
It is a severe, chronic condition that requires lifelong, intensive medical treatment to manage cholesterol levels and prevent fatal complications.










How It Affects You
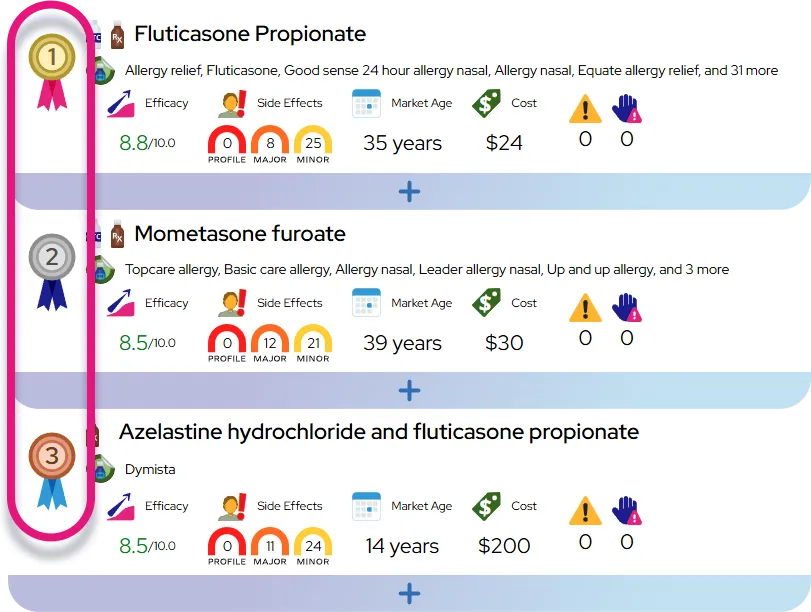
Homozygous familial hypercholesterolemia leads to extreme levels of cholesterol in the bloodstream from birth, causing fatty deposits to accumulate rapidly in various tissues. This buildup primarily damages the cardiovascular system by narrowing the arteries and stiffening heart valves, but it also creates visible signs on the skin and eyes. Key effects on the body include:
- Rapid development of plaque in the heart arteries and aorta, restricting blood flow.
- Visible, waxy cholesterol bumps on the hands, elbows, knees, buttocks, and ankles.
- Grayish-white cholesterol rings forming around the colored part of the eyes.
Causes and Risk Factors
Genetic Causes
Homozygous familial hypercholesterolemia is caused by inheriting two mutated copies of genes responsible for clearing cholesterol from the blood, one from each parent. Most commonly, this involves the LDL receptor gene, which normally acts like a claw to grab bad cholesterol and remove it from circulation. When a child inherits two defective copies, their liver cannot effectively remove low-density lipoprotein (LDL) cholesterol, causing it to build up to massive levels in the bloodstream from birth. Less frequently, mutations in other related genes can cause the same effect.
Risk Factors
The primary risk factor is a family history of Familial Hypercholesterolemia. Specifically, both parents usually have the heterozygous form of the condition, meaning they each carry one abnormal gene and likely have high cholesterol themselves. If both parents carry the trait, there is a 25 percent chance with each pregnancy that their child will inherit the homozygous (more severe) form. Ethnicity can sometimes be a factor, as the condition appears more frequently in populations with a "founder effect," where the gene mutation is more common within a specific group.
Prevention
Because this is a genetic disorder determined at conception, it cannot be prevented through lifestyle changes or vaccines. Primary prevention involves genetic counseling for prospective parents who have high cholesterol or a family history of early heart disease. Identifying carriers before they have children allows for informed family planning. Once a child is born with the condition, prevention shifts to stopping the progression of heart disease. This requires immediate and aggressive medical intervention to lower cholesterol levels and prevent early heart attacks.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms of homozygous familial hypercholesterolemia often appear in the first few years of life. The most distinct physical signs are xanthomas, which are waxy, orange or yellow deposits of cholesterol under the skin. These frequently appear on the hands (especially the webbing between fingers), elbows, knees, buttocks, and ankles. Children may also develop corneal arcus, a gray or white ring of cholesterol around the iris of the eye, which is highly unusual for young people. Internally, the most dangerous symptom is the rapid narrowing of arteries (atherosclerosis) and heart valves (aortic stenosis), which can lead to chest pain (angina), shortness of breath, or heart attacks even in childhood or adolescence.
Diagnostic Process
Clinicians usually suspect the condition based on the presence of visible xanthomas and extremely high cholesterol levels found during routine blood work or screening due to family history. A lipid panel will typically show LDL cholesterol levels above 400 mg/dL, and sometimes exceeding 1,000 mg/dL, which is several times higher than the normal range. Diagnosis is confirmed through genetic testing to identify the specific mutations inherited from both parents. Doctors also use imaging tests, such as echocardiograms or CT scans, to check for silent heart disease or blockages in the aorta and coronary arteries.
Treatment and Management
Medications and Procedures
Treatment must be aggressive and start as early as possible. Standard cholesterol-lowering drugs like high-intensity statins and ezetimibe are used, but they are often not powerful enough on their own because they rely on the liver receptors that are defective in these patients. Therefore, specialized medications such as PCSK9 inhibitors, evinacumab, or lomitapide are frequently prescribed to lower LDL levels through different mechanisms. A standard procedure for many patients is lipoprotein apheresis, a dialysis-like treatment where the patient's blood is passed through a machine to physically filter out the LDL cholesterol. This is typically done every one to two weeks. In very severe cases where other treatments fail, a liver transplant may be considered, as a healthy liver provides functional receptors to clear cholesterol.
Lifestyle Management
While diet and exercise alone cannot control this condition, a heart-healthy lifestyle remains crucial to minimize additional risks. Patients are advised to eat a diet low in saturated and trans fats, avoid smoking entirely, and maintain regular physical activity as approved by a cardiologist. Maintaining a healthy weight and managing other heart risk factors like blood pressure is essential to protecting the cardiovascular system.
When to Seek Medical Care
Patients require frequent, routine follow-up with a lipid specialist and cardiologist to monitor cholesterol levels and heart function. Emergency medical care should be sought immediately if a person experiences symptoms of a heart attack, such as sudden chest pain or pressure, pain spreading to the arm or jaw, difficulty breathing, or sudden fainting. Parents should contact their provider if they notice new or growing skin bumps (xanthomas) on their child or if the child complains of chest discomfort during physical play.
Severity and Prognosis
Severity and Complications
Homozygous familial hypercholesterolemia is the most severe form of hereditary high cholesterol. Without treatment, the arteries become severely blocked by adolescence. The major complication is premature cardiovascular disease, including narrowing of the aortic valve (supravalvular aortic stenosis) and coronary artery disease. This puts patients at extremely high risk for fatal heart attacks or strokes in their teenage years or early twenties. The condition is progressive, meaning the longer cholesterol levels remain high, the more damage occurs to the blood vessels.
Prognosis
Historically, the life expectancy for untreated patients was very short, with many not surviving past age 30. However, the prognosis has improved dramatically with the advent of modern therapies like apheresis and new pharmaceutical agents. When diagnosed early and treated aggressively, patients can significantly delay the onset of heart disease and live well into adulthood. The outcome depends heavily on how much the LDL cholesterol can be lowered and how early treatment begins. Regular monitoring is vital to detect and treat heart issues before they become critical.
Impact on Daily Life
Daily Impact and Coping
Living with this condition requires a significant commitment to medical routines. For many, this involves weekly or biweekly trips to a hospital for apheresis treatments, which can take several hours and affect school or work schedules. Taking multiple daily medications and navigating potential side effects is also part of the routine. The visible signs of the disease, like skin bumps, can sometimes affect self-esteem or social interactions in children. Families often deal with anxiety regarding the child's heart health, leading to hyper-vigilance about chest pain or physical exertion. Support groups and counseling can be valuable for managing the emotional burden of a chronic, life-threatening condition.
Questions to Ask Your Healthcare Provider
Being prepared for appointments helps ensure the best care. Consider asking the following questions:
- What are our specific target goals for LDL cholesterol levels?
- Am I (or is my child) a candidate for newer medications like evinacumab?
- How often do we need to schedule imaging tests to check heart health?
- Is lipoprotein apheresis necessary, and where is the nearest center?
- What specific physical activities are safe, and which should be avoided?
- Are there clinical trials available that we should consider?
- How should we screen other family members for this condition?
Common Questions and Answers
Q: Is homozygous familial hypercholesterolemia the same as just having high cholesterol?
A: No, it is much more severe. While "regular" high cholesterol is often caused by lifestyle and aging, this condition is a genetic disorder that causes extremely high cholesterol levels from birth, regardless of diet or exercise habits.
Q: Can a healthy diet cure this condition?
A: No. While a heart-healthy diet is important, it cannot cure the genetic defect. The liver simply cannot remove cholesterol from the blood, so medication and medical procedures are always necessary alongside dietary changes.
Q: Is it safe for children with this condition to play sports?
A: Physical activity is generally encouraged, but it depends on the severity of heart disease. A cardiologist must evaluate the child's heart function and provide specific guidelines on which activities are safe and which intense sports should be avoided.
Q: Will the skin bumps (xanthomas) ever go away?
A: Yes, in many cases, if cholesterol levels are lowered significantly and maintained at a lower level through treatment, the fatty deposits under the skin can shrink or disappear over time.
Q: Can people with this condition have children?
A: Yes, people with this condition can have children, but their children will inherit at least one copy of the gene (making them heterozygous). Genetic counseling is highly recommended to understand the risks and discuss family planning options.