
Late-onset Pompe disease
At a Glance
 It is a chronic, progressive condition that is treatable and manageable with lifelong therapy but currently has no cure.
It is a chronic, progressive condition that is treatable and manageable with lifelong therapy but currently has no cure.
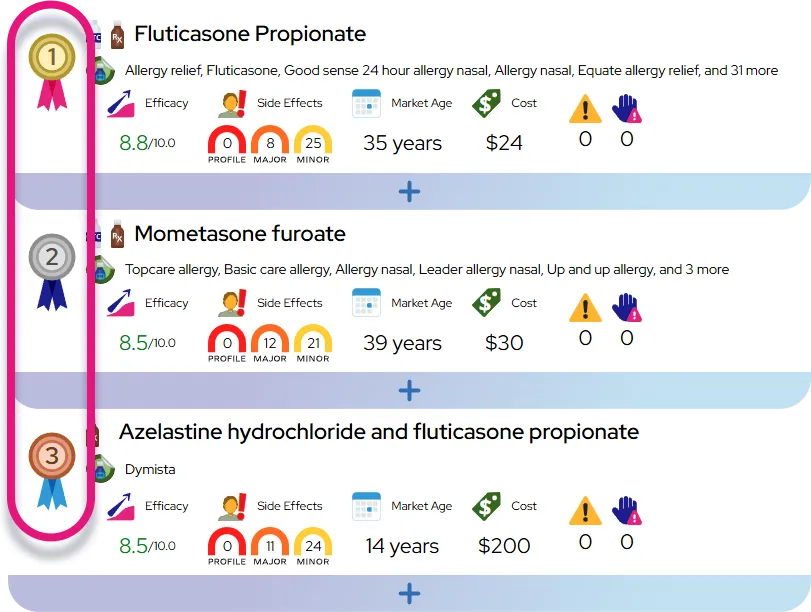










How It Affects You
Late-onset Pompe disease primarily affects the skeletal muscles closest to the center of the body (proximal muscles) and the respiratory muscles. A deficiency in the enzyme acid alpha-glucosidase leads to the toxic accumulation of glycogen within cells, causing progressive muscle damage and weakness. Key effects include:
- Muscle Weakness: Significant weakness in the hips, thighs, back, and shoulders, leading to difficulty walking or lifting arms.
- Respiratory Insufficiency: Weakness of the diaphragm and chest wall muscles, causing shortness of breath and sleep-disordered breathing.
- Mobility Challenges: A waddling gait and difficulty standing up from a seated position or climbing stairs due to pelvic and leg weakness.
Causes and Risk Factors
Underlying Biological Causes
Late-onset Pompe disease is caused by mutations in the GAA gene. This gene is responsible for producing an enzyme called acid alpha-glucosidase. Normally, this enzyme breaks down a complex sugar called glycogen within the lysosomes (recycling centers) of cells. In people with this condition, the enzyme is either missing or functioning poorly, causing glycogen to accumulate to toxic levels. This buildup primarily damages muscle cells, leading to progressive weakness and wasting.
Genetic Risk Factors
The primary risk factor is having parents who carry the gene mutation. The condition is inherited in an autosomal recessive pattern. This means an individual must inherit two copies of the mutated gene—one from each parent—to develop the disease. People who inherit only one copy are carriers and typically do not show symptoms, but they can pass the gene to their children.
Prevention and Screening
There is no known way to prevent the onset of the disease if a person has inherited the genetic mutations. However, for families with a known history of Pompe disease, genetic counseling and testing can help assess risks for future children. In some regions, newborn screening programs identify the condition early, allowing for monitoring and prompt treatment before significant muscle damage occurs. Secondary prevention strategies, such as starting treatment early, focus on preventing the progression of muscle weakness rather than preventing the disease itself.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms of late-onset Pompe disease can vary widely and may progress slowly over years. The most common early sign is proximal muscle weakness, affecting the hips, upper legs, and shoulders. This often results in a waddling gait, difficulty climbing stairs, or trouble standing up from a chair (Gowers' sign). Unlike the infantile form, heart involvement is rare. Respiratory symptoms are also a hallmark and may appear before significant muscle weakness. These include shortness of breath after physical effort, morning headaches, and daytime sleepiness caused by breathing difficulties during sleep (sleep apnea).
Diagnosis
Clinicians use several tools to identify this condition, which is often mistaken for other muscular dystrophies. The primary diagnostic test is an enzyme assay, often performed on a dried blood spot, to measure the activity of acid alpha-glucosidase. If enzyme activity is low, the diagnosis is confirmed with genetic testing to identify specific mutations in the GAA gene. Additional tests may include electromyography (EMG) to assess muscle electrical activity, muscle biopsies (less common now due to genetic testing), and pulmonary function tests to evaluate breathing strength, particularly when lying down.
Treatment and Management
Medical Treatments
The standard treatment for late-onset Pompe disease is Enzyme Replacement Therapy (ERT). This involves regular intravenous (IV) infusions of a laboratory-made version of the missing enzyme (such as alglucosidase alfa or avalglucosidase alfa). ERT helps break down the accumulated glycogen, which can slow the progression of muscle weakness and improve or stabilize respiratory function. Newer therapies, including combination treatments with enzyme stabilizers, are also becoming available to enhance the effectiveness of the enzyme.
Supportive Management
Management extends beyond medication to address daily function. Physical therapy is crucial to maintain range of motion and muscle strength without overexerting damaged muscles. Respiratory therapy involves monitoring lung function and using breathing support, such as BiPAP or CPAP machines, particularly at night. dietary adjustments, including a high-protein diet, may be recommended by some specialists to support muscle maintenance, though this should be supervised by a nutritionist.
When to Seek Medical Care
Regular follow-up with a specialist is essential to monitor disease progression. Patients should see a doctor immediately if they experience:
- Worsening Shortness of Breath: Difficulty breathing, especially when lying flat, or frequent respiratory infections.
- Signs of Respiratory Failure: Morning headaches, confusion, or extreme daytime fatigue.
- Mobility Changes: Increased falls or a sudden decline in the ability to walk or stand.
- Swallowing Difficulties: Choking or trouble eating, which can lead to weight loss and aspiration.
Severity and Prognosis
Severity and Progression
Late-onset Pompe disease is generally less severe than the infantile-onset form because some enzyme activity usually remains. However, it is still a serious, progressive disorder. The rate of decline varies significantly from person to person. Some individuals experience a very slow course with mild weakness appearing in adulthood, while others may have a more rapid progression starting in childhood. The disease primarily targets skeletal and respiratory muscles, leading to loss of mobility and lung function over time.
Prognosis
The long-term outlook has improved significantly with the advent of Enzyme Replacement Therapy (ERT). Before treatment was available, respiratory failure was the leading cause of reduced life expectancy. With modern treatment, disease progression can often be slowed, and many patients live well into adulthood. However, long-term complications such as permanent muscle weakness, scoliosis (curvature of the spine), and the need for wheelchair assistance or nighttime ventilation remain possible. Early diagnosis and treatment are the most critical factors in improving the prognosis.
Impact on Daily Life
Impact on Daily Activities
Living with late-onset Pompe disease requires adapting to physical limitations. Fatigue and muscle weakness can make daily tasks like grocery shopping, housekeeping, or working a full day challenging. Many individuals use energy conservation techniques, such as pacing activities and using assistive devices like scooters or raised toilet seats, to maintain independence. Respiratory weakness may require the use of a ventilator mask at night, which can impact sleep quality but is vital for energy levels the next day. The emotional toll of a chronic, progressive illness can also lead to anxiety or depression, making mental health support an important part of care.
Questions to Ask Your Healthcare Provider
Patients can better manage their condition by asking focused questions during appointments:
- What is my current baseline for muscle and lung function, and how often will we monitor it?
- Am I a candidate for the newest forms of enzyme replacement therapy?
- What specific physical therapy exercises are safe and effective for my condition?
- Should I see a dietician to discuss protein intake or weight management?
- How can I tell the difference between normal fatigue and disease progression?
- Are there patient advocacy groups or support networks you recommend?
Common Questions and Answers
Q: Is late-onset Pompe disease fatal?
A: It is not immediately fatal, but it is a progressive condition that can shorten life expectancy if untreated, primarily due to respiratory failure. Modern treatments have significantly improved survival rates and quality of life.
Q: How is it different from muscular dystrophy?
A: While both conditions cause muscle weakness, Pompe disease is a metabolic disorder caused by an enzyme deficiency and glycogen buildup, whereas muscular dystrophies are caused by defects in proteins that build muscle structure. Pompe disease is treated with enzyme replacement, while most muscular dystrophies currently lack specific treatments.
Q: Can exercise help with this condition?
A: Yes, moderate, supervised exercise is generally recommended to maintain muscle tone and flexibility. However, strenuous exercise can potentially damage weakened muscles, so a physical therapist should design a safe program.
Q: Will my children inherit the disease?
A: Because it is an autosomal recessive disorder, your children will only develop the disease if your partner also carries a mutation in the same gene. If your partner is not a carrier, your children will be carriers (like you) but will not have the disease. Genetic counseling can provide specific answers for your family.