
Phenylketonuria
At a Glance
 It is a chronic, lifelong condition that is not curable but is highly treatable and manageable through strict dietary control and medication.
It is a chronic, lifelong condition that is not curable but is highly treatable and manageable through strict dietary control and medication.










How It Affects You
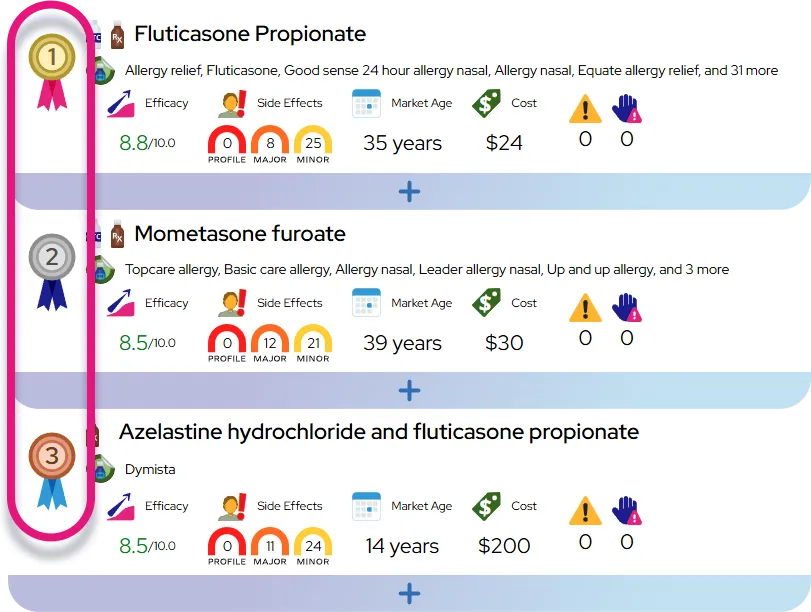
Phenylketonuria (PKU) is a rare metabolic disorder that prevents the body from breaking down an amino acid called phenylalanine, which leads to a harmful buildup of this substance in the blood and brain. If left untreated, high levels of phenylalanine can damage the central nervous system and interfere with normal growth and development. The effects on the body include:
- Toxic accumulation in the brain causing intellectual disability, developmental delays, and seizures.
- Reduced melanin production leading to lighter skin, hair, and eye color than family members.
- Systemic signs such as skin rashes (eczema) and a musty odor in the breath, skin, or urine.
Causes and Risk Factors
Causes of Phenylketonuria
Phenylketonuria is caused by a genetic mutation in the PAH gene. This gene provides instructions for making an enzyme called phenylalanine hydroxylase. In a healthy body, this enzyme converts phenylalanine (an amino acid found in all proteins and some artificial sweeteners) into other useful substances. People with this condition do not have enough of this enzyme, or it does not work correctly. As a result, when a person eats foods containing protein, phenylalanine builds up in the body to dangerous levels.
Risk Factors
The primary risk factor is genetics. The condition is inherited in an autosomal recessive pattern. This means a child must inherit two copies of the mutated gene, one from each parent, to develop the condition. Parents who each carry one copy of the mutated gene are called carriers; they typically do not show symptoms themselves but have a 25 percent chance with each pregnancy of having a child with the condition. Risk is also higher in people of certain ethnic ancestries, such as those of Northern European or Native American descent, though it occurs worldwide.
Prevention
Because the condition is genetic, there is no way to prevent the mutation from occurring. However, secondary prevention focuses on preventing the damage caused by the condition. This is achieved through universal newborn screening, which identifies affected infants immediately after birth. Early identification allows for immediate dietary intervention, which prevents brain damage and developmental issues before they start.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Newborns with phenylketonuria typically do not show symptoms at birth. Without treatment, symptoms usually appear within a few months as phenylalanine accumulates in the body. Early signs may include a musty odor in the breath, skin, or urine, which is caused by the excess byproduct of phenylalanine. Other symptoms can include skin rashes like eczema, vomiting, and lighter skin, eyes, and hair compared to family members. If the condition remains untreated, symptoms become more severe and may result in irreversible brain damage, marked by intellectual disabilities, seizures, delayed development, behavioral problems, and hyperactivity.
How Phenylketonuria Is Diagnosed
Diagnosis almost always occurs through newborn screening programs. A few drops of blood are taken from a newborn's heel one to two days after birth and tested for high levels of phenylalanine. If the screening test is positive, additional blood and urine tests are performed to confirm the diagnosis and determine the specific type of the condition. Genetic testing may also be done to identify the specific gene mutations. Prenatal testing is possible for families with a known history of the condition.
Differential Diagnosis
Clinicians may need to rule out other causes of hyperphenylalaninemia (high phenylalanine). This includes tetrahydrobiopterin (BH4) deficiency, a rarer condition where the body lacks a cofactor needed for the enzyme to work. Distinguishing between these conditions is vital because the treatment approaches differ.
Treatment and Management
Dietary Management
The main treatment for phenylketonuria is a strict, lifelong low-protein diet. Because phenylalanine is found in almost all proteins, people with this condition must avoid high-protein foods such as milk, dairy products, meat, fish, chicken, eggs, beans, and nuts. They must also avoid aspartame, an artificial sweetener that contains phenylalanine. To ensure the body gets enough nutrition, individuals consume a special metabolic formula or medical food that provides all necessary amino acids except phenylalanine. This diet typically begins immediately after diagnosis in infancy and should continue for life.
Medications
Some individuals may benefit from medication in addition to diet. Sapropterin is a drug that helps lower phenylalanine levels in people with certain forms of the condition who respond to it. Another option for adults is enzyme substitution therapy, which involves injections of an enzyme that breaks down phenylalanine. These treatments do not always replace the need for dietary restrictions but allows for a somewhat more flexible diet.
Monitoring and Follow-Up
Regular blood tests are essential to monitor phenylalanine levels. Frequency of testing varies by age and stability of the condition, ranging from weekly in infants to monthly in adults. Keeping levels within a safe range is critical to protecting brain function.
When to Seek Medical Care
Patients should see a doctor if they experience behavioral changes, difficulty concentrating, or mood shifts, as these may indicate high phenylalanine levels. Emergency care is rarely needed for the condition itself but may be required for severe seizures. Routine follow-up is necessary throughout life, and it is especially critical for women planning to become pregnant. High phenylalanine levels during pregnancy can severely harm the unborn baby, so strict control is required before conception and throughout pregnancy.
Severity and Prognosis
Severity Levels
The severity of phenylketonuria depends on how much functional enzyme is produced. In "classic PKU," the enzyme is completely absent or severely deficient, leading to very high phenylalanine levels and a high risk of brain damage without treatment. Mild or moderate forms involve some enzyme activity, resulting in lower phenylalanine levels and a less restrictive diet, though monitoring is still required.
Prognosis and Life Expectancy
When diagnosed early and treated consistently, the prognosis is excellent. Individuals can expect a normal life expectancy and normal intelligence. However, if treatment is stopped or not followed strictly, phenylalanine levels can rise, leading to lower IQ, memory loss, concentration problems, and mood disorders like anxiety or depression. Adults who return to the diet often see improvements in these symptoms.
Complications
The most serious complications occur in untreated individuals and include severe intellectual disability and neurological problems. A specific concern is "Maternal PKU syndrome." If a woman with the condition has high phenylalanine levels during pregnancy, her baby is at high risk for birth defects, including small head size (microcephaly), heart defects, and intellectual disability, even if the baby does not inherit the condition itself. Strict dietary control prevents these complications.
Impact on Daily Life
Impact on Daily Activities
Living with phenylketonuria requires constant attention to diet. Daily life involves weighing food, counting grams of protein, and preparing special medical formulas. This can make social activities involving food, such as parties or dining out, challenging. Children may feel different from their peers because they cannot eat common foods like pizza or ice cream. Planning ahead is essential for school lunches, travel, and events.
Mental and Emotional Health
Adhering to a restrictive diet can be stressful and may lead to feelings of isolation or burnout. Support groups and counseling can help patients and families cope with the emotional burden. Cognitive issues, such as slower processing speed or difficulty with executive function, may occur even in treated individuals, requiring accommodations at school or work.
Questions to Ask Your Healthcare Provider
Patients or parents should ask specific questions to manage the condition effectively:
- What is my (or my child's) current target range for phenylalanine levels?
- How often do we need to draw blood for monitoring?
- Are there new foods or medical formulas available that might taste better or be easier to prepare?
- Am I a candidate for medications that might allow for more protein in my diet?
- What specific resources or support groups do you recommend for families with this condition?
- For women: What steps must I take before trying to become pregnant?
Common Questions and Answers
Q: Can people with phenylketonuria ever eat normal food?
A: Most people with the classic form must avoid high-protein foods like meat and dairy for their entire lives. However, they can eat many fruits, vegetables, and special low-protein breads and pastas. Some individuals with milder forms or those on medication may tolerate small amounts of natural protein.
Q: Is phenylketonuria curable?
A: There is currently no cure, but it is effectively manageable. Gene therapy is being researched as a potential future cure, but strict lifelong management remains the standard of care today.
Q: What happens if an adult stops the diet?
A: While the brain is fully developed in adulthood, high phenylalanine levels can still cause toxicity. Adults who stop the diet may experience "brain fog," slow reaction times, anxiety, depression, and memory problems. Returning to the diet typically improves these symptoms.
Q: Can a woman with the condition have healthy children?
A: Yes, women with the condition can have healthy babies, provided they maintain strict control of their phenylalanine levels before and during pregnancy. This usually requires very frequent monitoring and a rigorous diet to prevent harm to the developing baby.
Q: Why do babies with the condition need a special formula?
A: Regular breast milk and infant formula contain phenylalanine, which the baby cannot process. The special formula provides all the protein and nutrients the baby needs to grow without the harmful amino acid.