
Progressive familial intrahepatic cholestasis
At a Glance
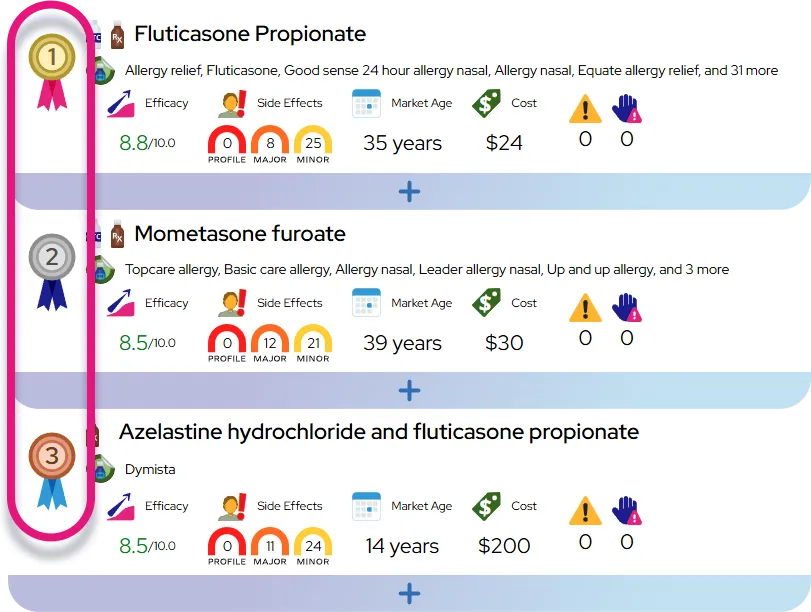
 PFIC is a chronic, progressive disease that requires lifelong management and often necessitates surgical intervention or liver transplantation.
PFIC is a chronic, progressive disease that requires lifelong management and often necessitates surgical intervention or liver transplantation.











How It Affects You
Progressive familial intrahepatic cholestasis primarily targets the liver by preventing the normal flow of bile, which leads to a toxic buildup of bile acids within liver cells. This accumulation causes progressive liver damage and scarring, but the effects are systemic and impact overall growth and comfort. Key effects on the body include:
- Intense, unrelenting itching (pruritus) that affects the skin across the entire body and disrupts sleep
- Yellowing of the skin and whites of the eyes (jaundice) due to high bilirubin levels
- Nutritional deficiencies and poor weight gain (failure to thrive) resulting from the body's inability to absorb fats and vitamins
Causes and Risk Factors
Genetic Causes
Progressive familial intrahepatic cholestasis is caused by gene mutations that impair the liver's ability to transport bile. In a healthy liver, specific proteins move bile acids and other bile components from liver cells into the bile ducts to aid digestion. In people with this condition, mutations in genes such as ATP8B1 (PFIC1), ABCB11 (PFIC2), and ABCB4 (PFIC3) result in missing or non-functioning transporter proteins. This defect causes bile to accumulate inside the liver cells (cholestasis) instead of flowing into the intestine, which leads to toxicity and tissue damage over time.
Inheritance and Risk Factors
The primary risk factor for developing this condition is having parents who both carry a gene mutation for the disease. It is inherited in an autosomal recessive pattern, meaning a child must inherit two defective copies of the gene—one from each parent—to develop the disorder. Parents who are carriers typically do not show symptoms themselves. If both parents are carriers, there is a 25% chance with each pregnancy that the child will have the condition. It affects males and females equally and can occur in any ethnic group.
Prevention Strategy
Because the condition is genetic, there are no lifestyle changes or vaccines that can prevent it from occurring. Primary prevention is limited to genetic counseling for families with a known history of the disease. For couples who are known carriers, genetic testing prior to or during pregnancy can identify if a child is at risk. Once diagnosed, management focuses on preventing complications rather than preventing the disease itself.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
The most distinct and debilitating symptom of this condition is severe itching (pruritus), which is often intense enough to cause skin damage from scratching and severe sleep disturbance. This itching typically starts in infancy. Other common signs include jaundice (yellowing of the skin and eyes), dark urine, and pale or clay-colored stools. Because bile is necessary for fat absorption, affected children often experience poor weight gain, slowed growth (failure to thrive), and deficiencies in fat-soluble vitamins (A, D, E, and K), which can lead to weak bones or bleeding problems. In some types of the disorder, such as PFIC1, patients may also experience extra-hepatic symptoms like watery diarrhea, hearing loss, or pancreatitis.
Diagnostic Tests
Clinicians suspect the condition based on the combination of cholestasis (bile buildup) and severe itching in a young child. Blood tests are the first step, revealing elevated total bile acids and bilirubin. A key differentiator in blood work is the Gamma-Glutamyl Transferase (GGT) level; it is typically low or normal in PFIC1 and PFIC2, which is unusual for liver disease, but elevated in PFIC3. To confirm the diagnosis, doctors perform genetic testing to identify the specific mutations. A liver biopsy may also be performed to examine tissue samples for specific patterns of scarring or transporter protein absence, helping to rule out other causes of liver disease.
Treatment and Management
Medications and Supplements
Medical management aims to reduce symptoms and improve growth. Doctors prescribe medications such as ursodeoxycholic acid (Ursodiol) to help move bile through the liver, though its effectiveness varies by subtype. To combat the severe itching, medications like rifampin, cholestyramine, or antihistamines are used. Recently, a new class of drugs called IBAT inhibitors (such as odevixibat) has been approved to treat pruritus in certain types of the condition by preventing bile acids from being reabsorbed in the intestine. Because patients cannot absorb fat well, high-calorie diets containing Medium-Chain Triglycerides (MCT oil) and supplements of fat-soluble vitamins (A, D, E, K) are standard to prevent malnutrition and bone weakness.
Procedures and Surgery
If medications do not sufficiently control itching or slow liver damage, surgical biliary diversion may be recommended. This procedure creates a pathway (stoma) from the gallbladder or intestine to the outside of the skin, allowing bile to drain into a bag rather than reabsorbing into the blood. This can significantly relieve itching and slow disease progression. For children with end-stage liver disease, liver failure, or suspected liver cancer (which is a risk in PFIC2), a liver transplant is the curative treatment. Transplantation replaces the defective liver with a healthy one, resolving the liver-based metabolic defect.
When to Seek Medical Care
Parents should seek prompt medical attention if an infant shows signs of persistent jaundice, pale stools, or fails to gain weight. Emergency care is needed if a child experiences signs of liver failure, such as confusion, excessive bleeding (like nosebleeds that won't stop), or vomiting blood. Routine follow-up is essential to monitor liver function, growth, and vitamin levels. Additionally, any sudden worsening of itching or new abdominal pain should be evaluated by a specialist.
Severity and Prognosis
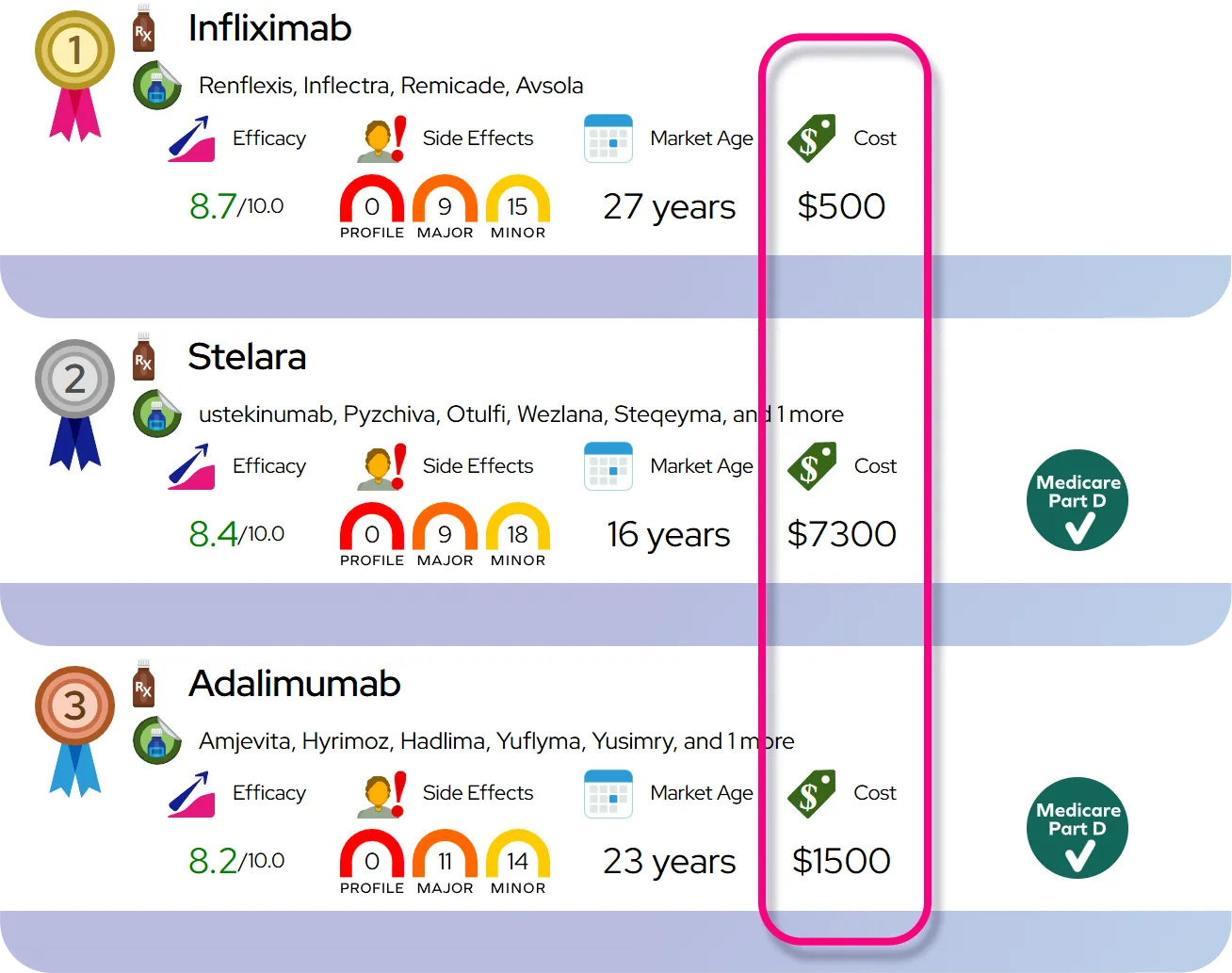
Severity and Disease Course
Progressive familial intrahepatic cholestasis is a severe condition that typically progresses to cirrhosis and end-stage liver disease within the first decade of life if left untreated. The rate of progression varies by genetic subtype. PFIC2 is often the most aggressive, with a rapid onset of liver failure and an increased risk of developing hepatocellular carcinoma (liver cancer) in early childhood. PFIC1 progresses somewhat more slowly but can be burdensome due to symptoms affecting other organs, such as the ears and pancreas. PFIC3 may present later in childhood or even adolescence and can vary from mild to severe.
Prognosis and Complications
Without treatment, the prognosis is poor, and mortality is associated with liver failure or bleeding complications. However, surgical interventions like biliary diversion can delay the need for a transplant by years. Liver transplantation is highly successful and generally provides an excellent long-term outlook, effectively curing the liver component of the disease. For PFIC1 patients, extra-hepatic symptoms like diarrhea may persist or worsen even after a transplant because the genetic defect exists in other tissues. Long-term risks include vitamin deficiencies leading to rickets or clotting disorders, and the complications associated with long-term immunosuppression after a transplant.
Impact on Daily Life
Managing Daily Activities and Coping
The most challenging aspect of daily life for families is often managing the intense itching, which can be distressing and disruptive. Children may scratch until they bleed, leading to skin infections and scarring. Keeping fingernails short, using soft bedding, and applying cooling moisturizers can provide some relief. Sleep disruption is common due to the itch, affecting the entire family's rest and the child's school performance. A specialized diet is also a major part of the daily routine, requiring strict adherence to vitamin supplements and special formulas containing MCT oil to ensure proper growth. Families often benefit from connecting with support groups to share coping strategies for the emotional and practical burden of a chronic rare disease.
Questions to Ask Your Healthcare Provider
- What specific genetic subtype does my child have, and how does that affect the treatment plan?
- Are there any new medications, such as IBAT inhibitors, that might help manage the itching?
- How often do we need to check vitamin levels and liver function?
- What are the signs that the liver disease is progressing or that a transplant might be needed?
- Are there dietary changes or specific formulas that will help with weight gain?
- What is the long-term plan for monitoring liver cancer risk?
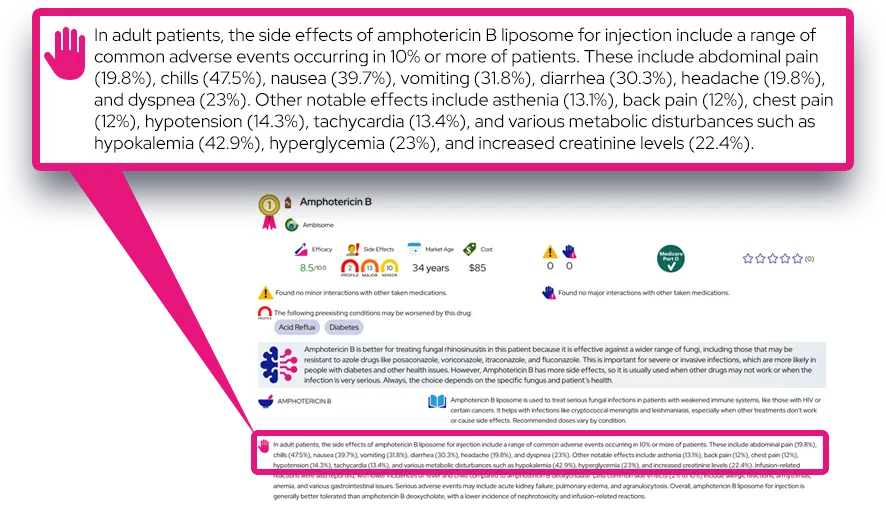
Common Questions and Answers
Q: Is there a cure for this condition?
A: There is no cure for the genetic defect itself, but a liver transplant is considered curative for the liver disease. It replaces the organ with one that functions normally, resolving the bile buildup and itching, although it requires lifelong anti-rejection medication.
Q: Can the condition be managed without surgery?
A: In some cases, medication and diet can manage symptoms and growth for a period of time. However, because the disease is progressive, many children eventually require a surgical biliary diversion or a liver transplant to prevent liver failure.
Q: Is the itching contagious or caused by an allergy?
A: No, the itching is not contagious and is not an allergic reaction. It is caused internally by the buildup of bile acids in the blood stream irritating the nerves, which is why standard allergy creams often do not work well.
Q: Will my future children also have this condition?
A: Because it is a recessive genetic disorder, there is a 25% chance in each pregnancy that another child will be born with the condition if both parents are carriers. Genetic counseling can provide specific risk assessments and testing options for future pregnancies.
Q: Does the condition affect intelligence or brain development?
A: The condition itself does not directly damage the brain or intelligence. However, severe nutritional deficiencies or chronic sleep deprivation from itching can impact a child's development and school performance if not well managed.