
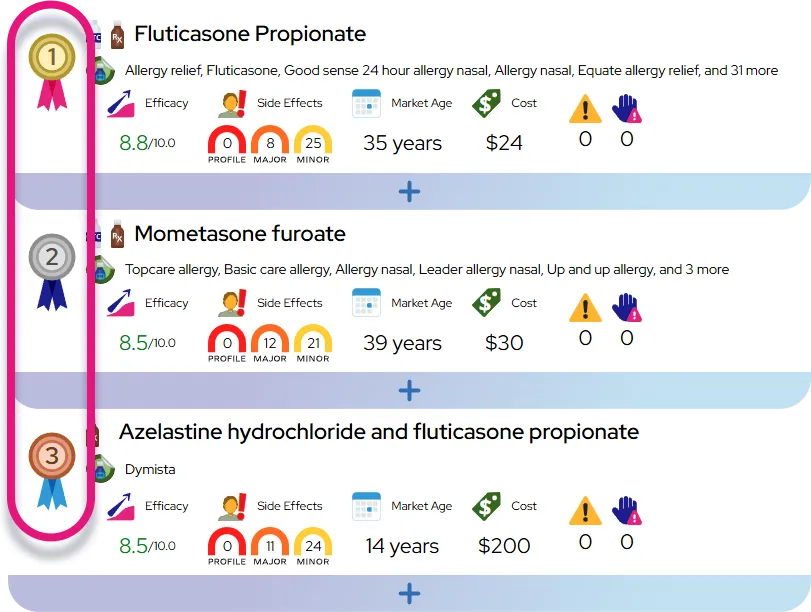
Sickle cell disease
At a Glance
 This condition is a chronic, lifelong illness that requires ongoing management but is characterized by acute episodes of severe pain and sudden health complications.
This condition is a chronic, lifelong illness that requires ongoing management but is characterized by acute episodes of severe pain and sudden health complications.




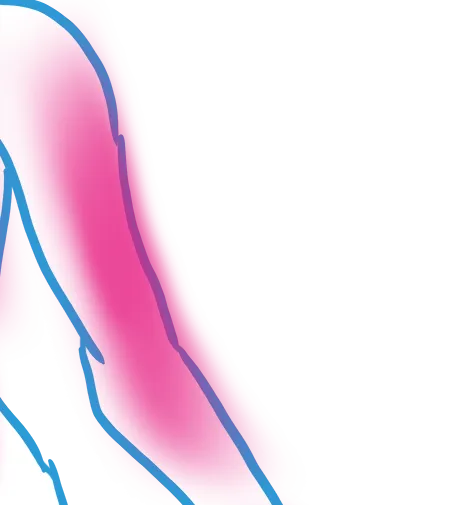






How It Affects You
Sickle cell disease is a systemic condition that affects the entire body because the abnormal, crescent-shaped red blood cells become rigid and sticky, causing them to get stuck in blood vessels and block blood flow to organs and tissues. This widespread obstruction prevents oxygen from reaching vital parts of the body, leading to episodes of severe pain and gradual damage to organs such as the spleen, liver, kidneys, lungs, and brain. Additionally, the rapid breakdown of these fragile cells results in chronic anemia and a weakened immune system, making the body more susceptible to infections and fatigue.
Causes and Risk Factors
Causes
Sickle cell disease is caused by a genetic mutation that affects hemoglobin, the protein in red blood cells that carries oxygen to the body. People with this condition inherit two faulty hemoglobin genes, one from each parent. This mutation causes red blood cells to lose their normal round, flexible shape and become rigid, sticky, and sickle-shaped. These abnormal cells die early, causing a constant shortage of red blood cells (anemia), and they clog small blood vessels, which cuts off blood flow and oxygen to tissues.
Risk Factors
The primary risk factor is genetics; a child must inherit the sickle cell gene from both parents to develop the disease. If a child inherits the gene from only one parent, they have sickle cell trait, meaning they generally do not have symptoms but can pass the gene to their children. The condition is most common in people whose ancestors come from sub-Saharan Africa, Spanish-speaking regions in the Western Hemisphere (South America, the Caribbean, and Central America), Saudi Arabia, India, and Mediterranean countries.
Prevention
Because sickle cell disease is an inherited genetic disorder, there is no way to prevent it if a person is born with it. Genetic counseling can help prospective parents understand their risk of passing the gene to their children. For those living with the disease, prevention focuses on avoiding pain crises and complications (secondary prevention). Strategies to reduce flare-ups include:
- Drinking plenty of water to avoid dehydration.
- Avoiding extreme temperatures, such as excessive cold or heat.
- Managing stress and getting adequate rest.
- Avoiding high altitudes where oxygen levels are low.
- Taking prescribed medications, such as hydroxyurea, to reduce the frequency of pain episodes.
- Staying up to date with vaccinations to prevent serious infections.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms of sickle cell disease can vary significantly from person to person and change over time. Signs usually appear around 5 to 6 months of age. Early symptoms often include painful swelling of the hands and feet (dactylitis) and fussiness from anemia. Common symptoms throughout life include:
- Anemia: Causing fatigue, paleness, and shortness of breath.
- Pain Crises: Sudden episodes of severe pain in the chest, abdomen, joints, or bones.
- Frequent Infections: Due to spleen damage which weakens the immune system.
- Delayed Growth: Slower growth and delayed puberty in children.
- Vision Problems: Caused by damage to the retina.
- Jaundice: Yellowing of the skin or whites of the eyes.
Diagnosis
In the United States and many other countries, sickle cell disease is identified through routine newborn screening using a blood test from a heel prick. If the screening is positive, further blood tests called hemoglobin electrophoresis are performed to confirm the diagnosis and determine the specific type of sickle cell disease. For older children or adults, blood tests can check for the defective form of hemoglobin.
Differential Diagnosis
Clinicians may need to distinguish sickle cell disease from other conditions that cause anemia or pain. These can include iron deficiency anemia, thalassemia (another blood disorder), leukemia, or other causes of acute abdominal or joint pain. Genetic testing and looking at the blood cells under a microscope help confirm the correct diagnosis.
Treatment and Management
Treatment Options
Treatment for sickle cell disease focuses on relieving pain, preventing infections, and managing complications. Modern treatments have significantly improved health outcomes. Common medications include:
- Hydroxyurea: Helps reduce the frequency of pain crises and the need for blood transfusions.
- L-glutamine: An oral powder that helps reduce acute complications.
- Voxelotor: Helps stop red blood cells from forming the sickle shape and improves anemia.
- Crizanlizumab: An intravenous medicine that reduces the frequency of pain crises by preventing cells from sticking to blood vessel walls.
- Pain Relievers: Over-the-counter medicines (like ibuprofen) or prescription opioids are used to manage pain episodes.
- Antibiotics: Daily penicillin is often prescribed for young children to prevent life-threatening infections.
Procedures and Potential Cures
Blood transfusions are commonly used to treat severe anemia or prevent stroke. A bone marrow transplant (also called a stem cell transplant) can cure the disease, but it carries significant risks and requires a matched donor. Recently, the FDA has approved gene therapies that offer a potential cure by modifying a patient's own stem cells to produce healthy hemoglobin, though these are new and complex procedures.
Lifestyle and Management
Self-care is a critical part of management. Patients are advised to stay well-hydrated, eat a heart-healthy diet, exercise moderately, and avoid smoking. Regular check-ups are essential to monitor organ health.
When to See a Doctor
Immediate medical attention is often required for certain symptoms. You should seek emergency care if you experience:
- A fever greater than 101°F (38.3°C), as this can signal a serious infection.
- Severe pain that is not relieved by home treatment.
- Difficulty breathing or chest pain (signs of acute chest syndrome).
- Sudden weakness, numbness, or difficulty speaking (signs of stroke).
- A painful erection lasting more than four hours (priapism).
- Sudden vision changes.
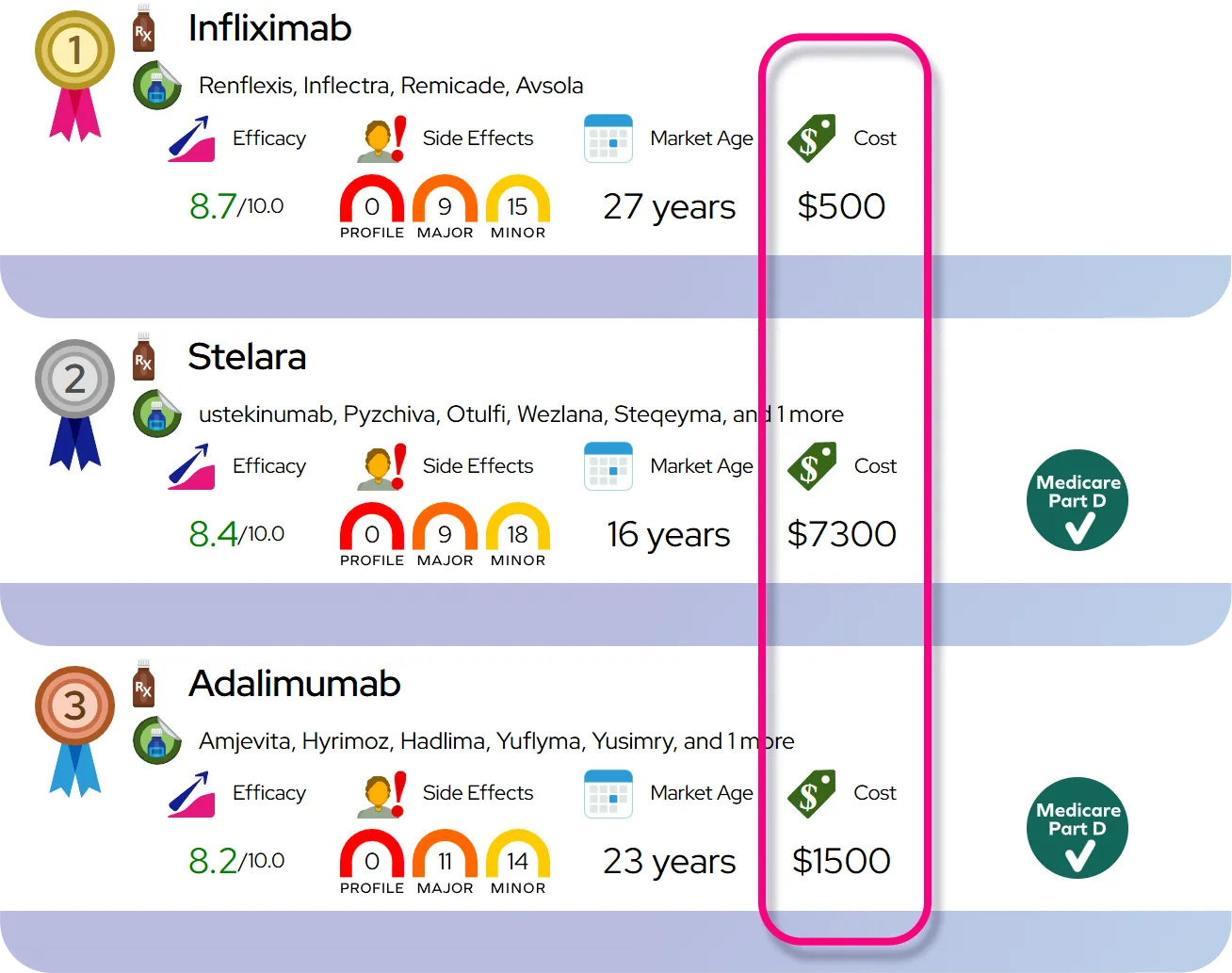
Severity and Prognosis
Severity
Sickle cell disease varies widely in severity. Some people have mild symptoms and infrequent pain episodes, while others experience severe, chronic pain and frequent hospitalizations. The type of sickle cell disease (such as HbSS versus HbSC) often influences severity, with HbSS generally being the most severe form. Factors that can worsen the condition include dehydration, infection, stress, and exposure to extreme temperatures.
Complications
Over time, the blockage of blood flow can damage organs throughout the body. Common complications include:
- Acute Chest Syndrome: A serious lung condition that causes chest pain, fever, and difficulty breathing.
- Stroke: Can occur in children and adults if blood flow to the brain is blocked.
- Organ Damage: Chronic oxygen deprivation can harm the kidneys, liver, and spleen.
- Leg Ulcers: Open sores on the lower legs that are difficult to heal.
- Pulmonary Hypertension: High blood pressure in the lungs, which shortness of breath.
Prognosis
In the past, many people with sickle cell disease did not survive childhood. Today, with early diagnosis and advanced care, the outlook has improved dramatically. People with the disease now often live into their 40s, 50s, and beyond. However, life expectancy is still shorter than that of the general population. Regular medical care, adherence to treatment plans, and early management of complications are the most important factors for a better prognosis.
Impact on Daily Life
Daily Activities
Living with sickle cell disease requires adjusting daily routines to avoid pain triggers. Fatigue from anemia can impact energy levels for school, work, or sports. Students may need special accommodations, such as permission to carry water bottles or take breaks during physical education. Adults may need flexibility in their work schedule to attend medical appointments or manage pain crises.
Mental and Emotional Health
Coping with a chronic, unpredictable painful condition can lead to feelings of anxiety, depression, or isolation. It is important to build a strong support system of family, friends, and support groups. Counseling or therapy can also be helpful in managing the emotional burden of the disease.
Questions to Ask Your Healthcare Provider
To better understand your condition and care plan, consider asking these questions at your next appointment:
- What specific type of sickle cell disease do I have, and what does that mean for my health?
- Am I a candidate for new treatments like gene therapy or a bone marrow transplant?
- What is my personal plan for managing pain at home versus going to the hospital?
- How often should I be screened for complications like stroke or eye damage?
- Are there any specific activities or dietary changes I should avoid?
- What vaccinations do I need to stay protected against infections?
- What are the risks of pregnancy, and how can they be managed?
Common Questions and Answers
Q: Is there a cure for sickle cell disease?
A: Yes, a bone marrow transplant can cure the disease for some patients, and recently approved gene therapies also offer a potential cure. However, these treatments involve significant risks and are not suitable for everyone.
Q: Is sickle cell disease contagious?
A: No, sickle cell disease is not contagious. It is a genetic condition inherited from your parents, so you cannot catch it from someone else like a cold or flu.
Q: Can a person with sickle cell disease have children?
A: Yes, women with sickle cell disease can have healthy pregnancies and healthy babies, but the pregnancy is considered high-risk and requires close monitoring by a specialist to manage potential complications.
Q: Does sickle cell trait make you sick?
A: Most people with sickle cell trait do not have symptoms and live normal lives. However, in rare cases, they may experience complications under extreme conditions, such as severe dehydration or very high physical exertion at high altitudes.
Q: Why do people with sickle cell disease need to drink so much water?
A: Dehydration causes red blood cells to become sticky and sickle-shaped more easily, which can trigger a pain crisis. Drinking plenty of water helps keep blood flow smooth and reduces the risk of these blockages.