
Tetrahydrobiopterin deficiency
At a Glance
 It is a chronic, lifelong condition that is treatable and manageable with continuous medication and monitoring.
It is a chronic, lifelong condition that is treatable and manageable with continuous medication and monitoring.










How It Affects You
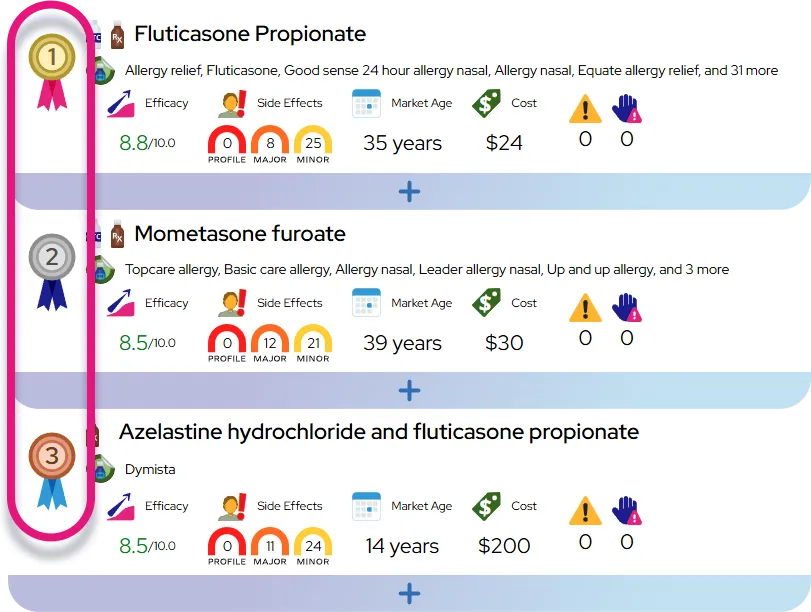
Tetrahydrobiopterin deficiency is a rare metabolic disorder that primarily affects the nervous system but can have widespread effects on muscle control and development throughout the body. This condition impairs the body's ability to process the amino acid phenylalanine and to produce essential neurotransmitters like dopamine and serotonin, which are critical for sending signals between nerve cells. Without proper management, toxic levels of phenylalanine build up in the blood while the brain is starved of chemical messengers, leading to progressive neurological issues.
Key effects on the body include:
- Neurological disruptions such as seizures, tremors, and difficulty regulating body temperature
- Movement disorders characterized by weak muscle tone (hypotonia) in the trunk and stiffness in the limbs
- Developmental delays affecting speech, motor skills, and intellectual growth
Causes and Risk Factors
Biological Causes
Tetrahydrobiopterin deficiency is caused by changes (mutations) in specific genes that control the production or recycling of tetrahydrobiopterin (BH4). BH4 is a cofactor, a helper molecule, that enzymes need to convert the amino acid phenylalanine into tyrosine. It is also essential for creating neurotransmitters such as dopamine, serotonin, norepinephrine, and epinephrine. When BH4 is missing or not working correctly, phenylalanine builds up in the body to toxic levels, and the levels of neurotransmitters fall dangerously low. This chemical imbalance disrupts normal brain function and development.
Genetic Risk Factors
The primary risk factor for this condition is family history, as it is an inherited genetic disorder. It typically follows an autosomal recessive pattern, meaning a child must inherit two copies of the non-working gene—one from each parent—to develop the condition. Parents who each carry one copy of the mutated gene generally do not show symptoms themselves but have a 25 percent chance of passing the condition to their children with each pregnancy. Rare forms may follow different inheritance patterns, but family genetics remains the central risk factor.
Prevention and Screening
There is currently no way to prevent the genetic mutations that cause tetrahydrobiopterin deficiency. Primary prevention focuses on genetic counseling for parents with a family history of the disorder, allowing them to understand their risks before starting a family. Secondary prevention relies heavily on newborn screening programs. Identifying the condition immediately after birth allows treatment to begin before significant brain damage occurs. Prenatal testing may also be available for families with known genetic risks.

Diagnosis, Signs, and Symptoms
Signs and Symptoms
Symptoms often appear within the first few months of life, although severity can vary depending on the specific gene affected. Early clinical signs may include low muscle tone (hypotonia) in the trunk of the body, combined with stiffness or spasticity in the arms and legs. Parents or clinicians may notice the infant is unusually lethargic, has difficulty feeding or swallowing, or fails to meet developmental milestones like holding their head up or sitting. As the condition progresses without treatment, symptoms can include seizures, tremors, uncontrolled muscle jerks, temperature instability, drooling, and microcephaly (an unusually small head size).
Diagnostic Tests
Diagnosis often begins with newborn screening, which checks for high levels of phenylalanine in the blood. Because high phenylalanine is also the hallmark of Phenylketonuria (PKU), infants with a positive result require further testing to distinguish BH4 deficiency from classic PKU. Doctors perform a specific BH4 loading test, where the baby is given a dose of BH4 to see if phenylalanine levels drop. Additional tests include analyzing urine for specific chemicals called pterins and measuring neurotransmitter metabolites in cerebrospinal fluid. Genetic testing is used to confirm the diagnosis by identifying the specific gene mutation involved.
Differential Diagnosis
This condition is most commonly confused with classic Phenylketonuria (PKU) because both present with high phenylalanine levels. However, standard PKU treatment (dietary restriction) does not improve the neurological symptoms of BH4 deficiency because it does not address the lack of neurotransmitters. It may also be initially mistaken for other causes of failure to thrive, cerebral palsy, or generalized dystonia due to the movement issues involved.
Treatment and Management
Medications and Therapies
The main goal of treatment is to normalize phenylalanine levels and restore the balance of neurotransmitters in the brain. Most patients require synthetic BH4 (sapropterin dihydrochloride) to lower phenylalanine levels. Unlike classic PKU, diet alone is rarely sufficient. Crucially, patients must take medication to replace the missing neurotransmitters. This typically involves taking L-dopa (often combined with carbidopa) to restore dopamine and 5-hydroxytryptophan (5-HTP) to restore serotonin. These medications often result in dramatic improvements in muscle tone, alertness, and development.
Dietary Management
While medication is central, some individuals may also need to follow a diet that limits phenylalanine intake to keep blood levels within a safe range. This diet is similar to the one used for PKU but is often less restrictive because the synthetic BH4 medication helps the body process some phenylalanine. A dietitian usually helps families plan meals that avoid high-protein foods like meat, eggs, and dairy if necessary, often supplementing with special medical formulas.
Monitoring and Follow-up
Management is lifelong and requires regular check-ups. Doctors monitor blood levels of phenylalanine and may test prolactin levels, which serve as a marker for dopamine status in the brain. Periodic adjustments to medication dosages are necessary as the child grows and gains weight. Developmental assessments help track cognitive and motor progress to ensure therapy is working effectively.
When to Seek Medical Care
Parents should seek immediate medical attention if their child experiences a seizure, breathing difficulties, or sudden changes in alertness. It is also important to contact a healthcare provider if there are signs of medication side effects, such as uncontrolled movements (dyskinesia) or gastrointestinal issues. Routine follow-up is essential if developmental milestones are missed or if symptoms like tremors or stiffness re-emerge, as this may indicate a need to adjust medication dosages.
Severity and Prognosis
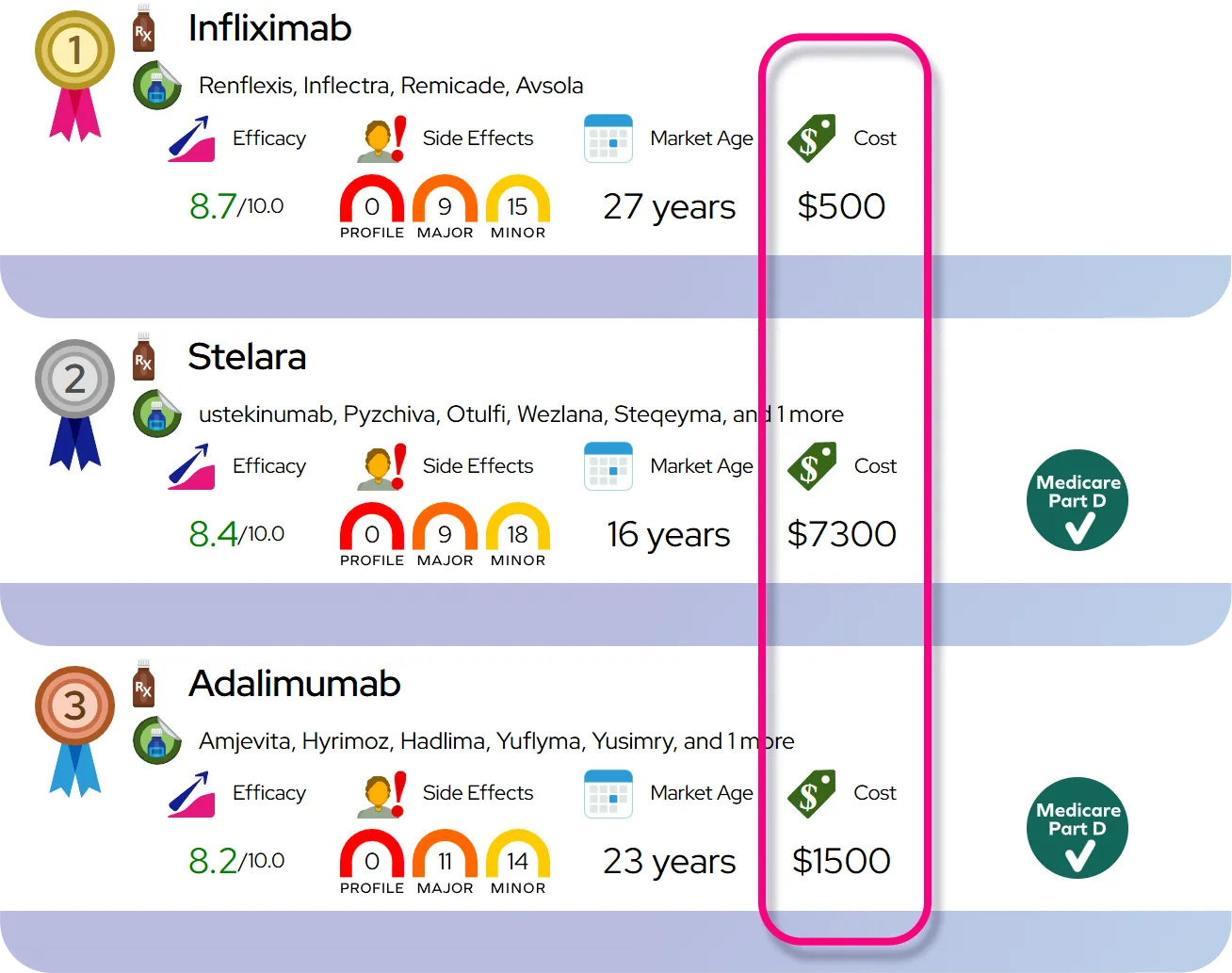
Severity and Disease Course
The severity of tetrahydrobiopterin deficiency varies widely depending on the specific enzyme defect and how early treatment begins. Without treatment, the condition is severe and progressive, leading to profound physical and intellectual disability. It is considered a chronic disorder that requires lifelong management. The course of the disease changes significantly with therapy; treated individuals often experience a halting of symptom progression and can achieve near-normal or normal functioning.
Potential Complications
If left untreated or poorly managed, complications can include severe intellectual disability, difficulty swallowing leading to aspiration pneumonia, and intractable seizures. Long-term risks involve orthopedic issues like scoliosis due to abnormal muscle tone. Overtreatment or improper balance of neurotransmitter replacement can cause temporary side effects like erratic movements (dyskinesia) or irritability, which resolve with dosage adjustments.
Prognosis
The prognosis is generally excellent for children diagnosed and treated shortly after birth. These individuals typically have normal intelligence and live independent lives. However, if treatment is delayed, existing neurological damage may not be fully reversible, although symptoms can still improve. Life expectancy is typically normal for well-managed patients, whereas untreated severe cases may have a shortened lifespan due to complications like infection or seizure-related injuries.
Impact on Daily Life
Daily Activities and Coping
Living with tetrahydrobiopterin deficiency involves a strict routine of medication administration, often multiple times a day. Families must be organized to ensure doses of L-dopa, 5-HTP, and sapropterin are never missed, as consistency is key to stability. School-aged children may need individualized education plans (IEPs) if they have experienced any developmental delays. Physical, occupational, and speech therapy are common parts of daily life in early childhood to maximize motor skills and communication. Emotional support for the family is important, as managing a rare chronic condition can be stressful.
Questions to Ask Your Healthcare Provider
- What specific gene mutation does my child have and how does it affect the treatment plan?
- How strictly do we need to control phenylalanine intake through diet versus medication?
- What are the signs that neurotransmitter medication dosages need to be adjusted?
- Are there any specific medications or foods we should strictly avoid?
- How often should we schedule blood tests and neurological assessments?
- What resources or support groups are available for families with this specific deficiency?
Common Questions and Answers
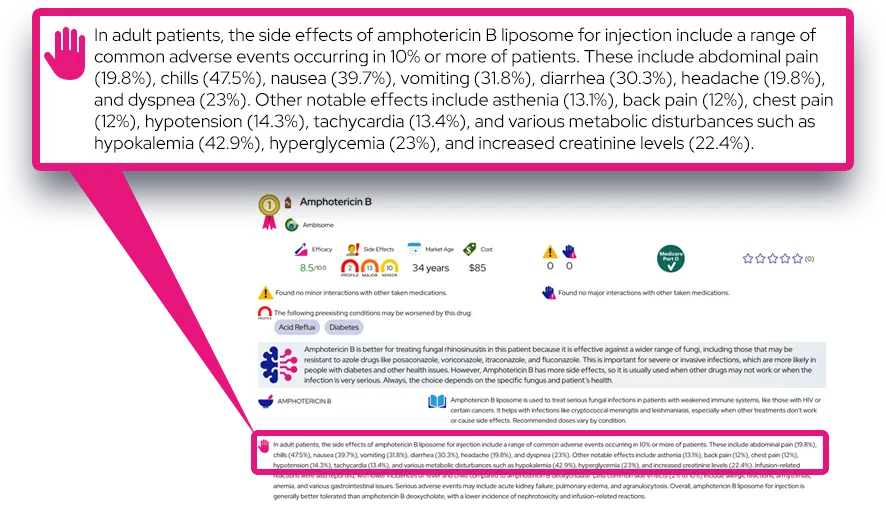
Q: Is tetrahydrobiopterin deficiency the same thing as PKU?
A: No, although both cause high phenylalanine levels. Classic PKU is an enzyme defect that only affects phenylalanine processing, while BH4 deficiency is a cofactor defect that also stops the production of essential neurotransmitters like dopamine and serotonin.
Q: Can a child outgrow this condition?
A: No, this is a lifelong genetic condition. The body will always lack the ability to naturally produce or recycle enough BH4, so medication and management are required throughout life.
Q: Is the damage reversible if treatment starts late?
A: Some symptoms can improve significantly with treatment, such as muscle tone and alertness. However, intellectual disability or structural brain damage that occurred before treatment began is usually permanent.
Q: Can women with this condition have healthy children?
A: Yes, with careful planning. Women with BH4 deficiency must maintain strict control of their phenylalanine levels and medication before and during pregnancy to prevent damage to the developing baby (maternal PKU syndrome).
Q: Why doesn't the PKU diet work by itself?
A: The PKU diet only lowers phenylalanine. It does not replace the missing neurotransmitters (dopamine and serotonin) that the brain needs to function, so neurological symptoms would continue without specific medication.